一种利用全基因组测序进行分离鉴定与精细定位水稻农艺性状基因的方法与流程
本发明涉及遗传育种
技术领域:
,特别涉及一种利用全基因组测序进行分离鉴定与精细定位水稻农艺性状基因的方法。
背景技术:
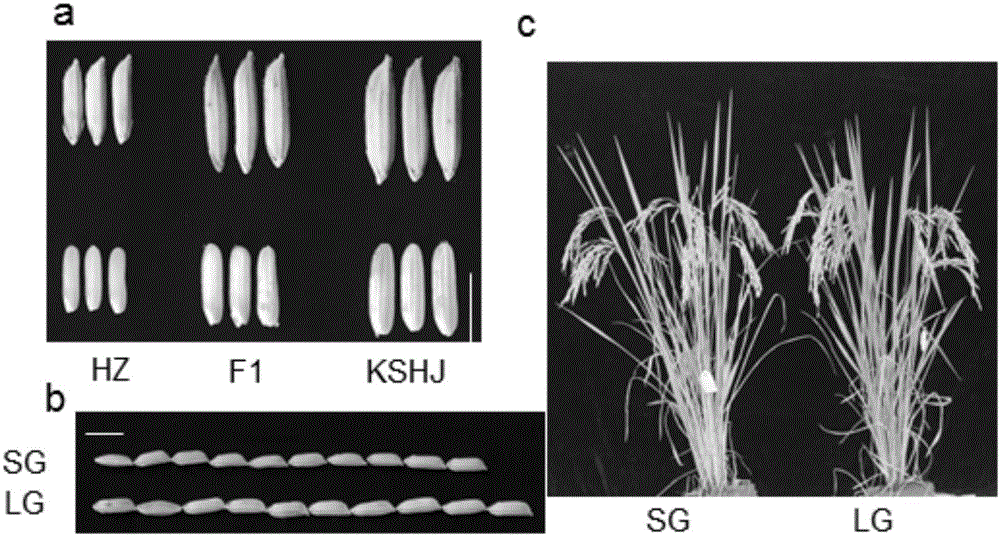
:在农业作物里,许多农艺性状都是由一个或多个基因调控的。尤其是一些重要农艺性状,往往都是由几个基因共同调控,这些基因被称之为数量性状位点(quantitativetraitloci,QTLs)。QTLs同样控制着许多水稻重要性状,尤其是一些产量相关的农艺性状。因此鉴定和分离QTLs一直是水稻研究的重点方向。目前,在水稻中已有许多QTLs被定位,但只有其中的一小部分被克隆,缺乏高效的QTLs克隆方法是其中一大制约因素。常规的QTLs克隆方法主要是图位克隆法,首先利用初级群体(F2或RIL)对目标性状的QTLs进行初步定位,确定主效QTL及其两侧分子标记;利用回交并结合分子标记辅助选择获得遗传背景单一且表型稳定的高世代回交后代(BCnF1),再筛选目标区域杂合的近等基因系,构建NIL-F2群体进行精细定位,获得候选基因。在这一过程中获得遗传稳定的回交后代及双亲在该QTL区间内的多态性是能否成功克隆到QTL的关键。传统的QTL定位方法费时费力,且对材料间的多态性要求甚高。随着DNA测序技术的发展,全基因组测序(Whole-genomesequencing,WGS)越来越多地被应用到植物基因的定位与克隆当中。WGS首先是被用于分析基因组间的遗传多态性,随后通过测序获得的丰富的遗传多样性被应用于全基因组关联分析(genome-wideassociationstudies,GWAS)。随着一些模式植物的基因组序列被组装完成后,以这些基因组为参照序列的第二代测序技术(next-generationsequencing,NGS)在基因的定位与克隆,尤其是QTLs的发掘上发挥了重要作用。测序获得的高密度遗传多态性大大提高了QTLs定位的精度。利用NGS结合分离群体分组分析法(Bulked-segregantanalysis,BSA)方法,已分离鉴定多个拟南芥突变体基因和一些水稻的QTLs。而在此基础上发展的QTL-seq技术,更为QTLs的快速准确定位提供了新的方法。技术实现要素:本发明的目的在于提供一种利用全基因组测序进行分离鉴定与精细定位水稻农艺性状基因的方法,该方法能快速准确的鉴定并精细定位水稻QTLs,为其进一步的克隆与功能分析打下坚实基础。本发明解决其技术问题所采用的技术方案是:一种利用全基因组测序进行分离鉴定与精细定位水稻农艺性状基因的方法,(1)以目标性状具有差异的两份水稻材料为亲本进行杂交,再以一方亲本为轮回亲本进行连续回交,获得目标性状分离、目标性状控制基因位点有且仅有杂合和轮回亲本两种基因型的3-6代回交F1群体;(2)选取3-6代回交F1群体中目标性状差异最大的两个单株分别进行全基因组重测序,或选取3-6代回交F1群体中与轮回亲本在目标性状上差异最大的单株及该轮回亲本分别进行全基因组重测序;(3)以区间内杂合态多态的分布频率为统计指标,对步骤(2)全基因组重测序结果中的杂合位点进行比较分析,获得两份水稻材料间在杂合位点分布频率具有显著差异的区段,选取携带目标性状的测序单株中的杂合位点频率显著大的区段为调控目标性状的候选基因区段;(4)参照步骤(3)序列分析结果,在调控目标性状的候选基因区段设计引物,再利用步骤(2)测序的回交F1群体中目标基因为杂合型的单株自交产生的F2群体作为图位克隆群体,对候选基因区段与目标性状间的连锁关系进行分析,对控制目标性状的基因位点进行分离鉴定与精细定位。作为优选,步骤(1)中的回交选择带有隐性性状一方亲本为轮回亲本进行回交,每代回交都选取带有显性性状的单株为父本。作为优选,步骤(4)中在调控目标性状的候选基因区段每1000kb-3000kb设计一对引物。本发明的有益效果是:方法不仅能快速准确的鉴定并精细定位水稻一个或多个主效数量性状基因,同时也可分离鉴定多个互作基因位点,为进一步的克隆与功能分析打下坚实基础。附图说明图1是本发明的技术路线图;图2是比较各世代材料的表型。a:水稻大粒品种KSHJ,小粒品种HZ及其F1的籽粒比较;b:BC4F1中大粒单株(LG)与小粒单株(SG)在粒型上的差异;c:SG与LG成熟期表型图。图3大粒材料与小粒材料在差异杂合位点分布频率的比较结果。图4大粒基因的关联分析及精细定位。图5BC4F1中光叶亲本与长茸毛单株测序结果杂合区段分析。图6叶毛发生相关基因LH5与LH6的定位。具体实施方式下面通过具体实施例,对本发明的技术方案作进一步的具体说明。本发明中,若非特指,所采用的原料和设备等均可从市场购得或是本领域常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。实施例:材料与方法:水稻材料:两组带有不同性状差异的水稻被用来验证试验方法。其中一组为粒型有明显差异的水稻品种况丝华巨(KSHJ,大粒籼稻品种)和华占(HZ,小粒籼稻品种),以华占为轮回亲本进行连续回交,获得BC4F1群体,从该群体中选取一大粒单株及一小粒单株作为测序样本;另一组为叶片表面带长茸毛的水稻籼稻品种菲B10和光叶美国稻品种Cypress,以Cypress为轮回亲本进行连续回交,获得BC4F1群体,从该群体中选取一叶表面长茸毛单株及轮回亲本Cypress作为测序样本。本发明使用的所有种质资源均由中国水稻研究所种质资源库提供。基因组DNA样品的备制及测序:四份测序样品的DNA用DNeasyPlantMiniKit(QIAGEN)提取,每份样品大约10ug高纯度DNA被用于后续测序。所有的测序工作和前期的数据预处理,包括去除无效数据,去除接头序列等均由杭州联川生物技术有限公司完成。序列的比对分析:以网上提供的水稻基因组序列为参照序列(http://ensembl.gramene.org/Oryza_indica/Info/Index和http://plants.ensembl.org/Oryza_sativa/Location/),将测序样本的序列分别与之进行比较,分别得到两者与参照序列具有差异的位点。对以上差异位点进行归类分析,可分为两者间相同或不同的差异位点。再选取两者特有的差异位点中的杂合位点进行分析,以区间内杂合态多态的在染色体上的分布频率为统计指标,比较两份样品的统计结果,获得两份水稻材料间在杂合位点分布频率具有显著差异的区段,将带有目标性状样品的杂合位点显著多于另一样本的染色体区段作为控制目标基因的候选区段。目标基因的定位分析:参照序列分析结果,在候选区段内设计引物,以BC4F1为定位群体,对检测到的差异序列与目标性状进行关联分析,确定目标基因所在的位点。以目标性状分离的BC4F2大群体为精细定位群体,对目标基因进行精细定位。结果与分析本发明的基本原理本发明中用到的定位与克隆水稻农艺性状基因的方法如图1所示,该技术结合了全基因组测序和传统QTLs定位克隆方法,可快速准确地定位水稻农艺性状基因,并为水稻农艺性状基因的克隆及功能分析打下深厚的材料基础。首先我们选取目标性状有差异的两份材料进行杂交,选择带有隐性性状一方亲本为轮回亲本进行回交,每代回交都选取带有显性性状的单株为父本。如果调控目标性状的基因为单个基因或一主效QTL,则每代回交F1中目标性状的分离比应该都接近于1:1,且在带有显性性状的单株中调控目标性状的DNA区域为杂合态,带有隐性性状的单株则为纯合态。因此,只需从回交F1中选取带有最大显性性状和隐性性状单株各一株进行基因组重测序。通过比较F1中的显性性状单株与隐性单株基因组中杂合态DNA的差异即能找出目标性状所在的基因位点。如果是多个基因调控目标性状,则在F1群体中选取目标性状表现最显著的一株,并选取轮回亲本作为比较样本。理论上杂交F1代所有的核染色体DNA都是杂合态的,即一半来自于母本,另一半来自于父本,每回交一代,核染色体DNA中杂合的就减少50%。回交4-5代后F1中杂合态的DNA是全基因组的6.25%-3.125%,以水稻基因组(430M)为例就只剩大约13.5M-27M的DNA序列为杂合态,其它区段都是纯合的且与轮回亲本一致,后续的定位工作只需分析这10%不到的基因组序列。从回交F1中选取带有最大显性性状和隐性性状单株或轮回亲本进行基因组重测序。为保证有足够的数据进行分析,对每个单株都进行10倍覆盖的测序。将测序结果与网上已有的参照基因组,例如OryzasativaJaponica(http://plants.ensembl.org/Oryza_sativa/Info/Index?db=core)进行比较,获得两份材料与参照基因组比较后在SNP和INDEL多态上的分布结果。统计出一定区间内杂合态多态的分布频率,比较两份材料在杂合态多态的分布频率上的差异,将比较所得的差异作为获选的目标基因,并参照测序结果在这些区域内设计引物,利用测序的显性回交单株的后代F2为图位克隆群体,对获选位点与目标性状间的连锁关系进行分析,获得目标性状基因并进一步精细定位该基因。对一水稻粒型主效基因的精细定位况丝华巨是一份大粒材料,其在粒长,粒宽及粒厚上均大于另一水稻经典籼稻材料华占,两者的杂交F1也明显大于华占(图2a)。以华占为轮回亲本,选取大粒性状的F1进行连续回交,获得BC4F1,从中选取一大粒单株(largegrainplant,LG)及一小粒单株(smallgrainplant,SG)作为测序样品,从表1中可知LG在粒长,粒宽,粒厚及百粒重等表型上与SG均有着显著的差异。在长宽厚上均有10%左右的增长,最终导致粒重有近30%的增长。但其他性状如株型等方面都极为相似(图2c)。表1BC4F1中大粒材料(LG)和小粒材料(SG)在粒型和粒重性状上的比较籽粒性状大粒材料(LG)小粒材料(SG)P值粒长(mm)10.55±0.189.36±0.274.28045E-22粒宽(mm)2.52±0.082.34±0.097.23077E-10粒厚(mm)2.11±0.081.93±0.12.09201E-08百粒重(g)2.93±0.052.24±0.043.59818E-08对大粒样本(LG)和小粒样本(SG)进行双端(PEpairedend)测序,其中大粒样本获得读长(reads)数目45776244条,共4577624400bp,其中高质量的数据(>Q20)3847153413bp,占总数84.04%;小粒样本(LG)获得读长(reads)数目45846502条,共4584650200bp,其中高质量的数据(>Q20)3810236166bp,占总数83.11%。应用滑动窗口法去除低质量片段和含N比例在10%以上的reads,最终LG获得(>Q30)3878195963bp,覆盖基因组10.4倍,SG获得(>Q30)3838631152bp,覆盖基因组10.3倍。表2多态统计表将两份材料序列分别与网上提供的籼稻基因组序列(ASM465v1)进行比较,获得两者与参照序列的差异位点。从表2我们可以看出,LG和SG与参照序列比对后获得的多态性都比较丰富,平均每kb就有1-2个多态;其中在LG里多态出现的最大间隔为131947bp,在SG里为125308bp。对两份材料的比对结果进行进一步分析,得到在两份材料里有差异的多态位点,这些差异位点包括只在一份材料里出现的多态位点或在一份材料里是杂合多态位点,而另一份材料里为纯合多态位点。从这些差异位点中选择测序深度大于5且AF值大于20%小于80%的位点作为可靠的杂合位点进行统计,从统计的表中我们也可以发现杂合态出现的频率一般为0.16左右,在第1,8,11号染色体两份样品的杂合频率明显高于其他染色体,推测这3条染色体两份材料都有杂合区域;而在第2染色体仅LG的杂合频率异常偏高,在第3、12染色体仅SG的杂合频率偏高,推测这三条染色体上仅一份材料出现杂合区域。为进一步鉴定出两份材料在基因组上的差别,我们统计了500kb区间杂合态多态出现的频率。结果表明如图3(和附图3对应吗,是的)所示,在染色体2,5,11各有一段区域是LG杂合频率远高于SG,由此推测在染色体2,5,11中表现出差异的区域很可能是大粒基因存在的位点,因此将基因组区段CHR2(chr2:18000000..33500000),CHR5(chr5:21000000..25000000)及CHR11(chr11:16500000-18500000)作为候选基因位点。以测序结果为依据,在候选基因区段设计INDEL引物,以SG和LG的基因组DNA为模板,利用LG的后代即BC4F2群体为QTL定位群体,对分析所得的三个候选位点每隔2000kb左右设计一对INDEL引物(表3),利用QTLIciMapping软件对这些标记与粒型的关联性进行分析。在chr2-2区间内我们选用了8对引物,在其他两个区间各选用2对引物,一共12对引物用作连锁分析。从BC4F2中随机选取76株作为QTL定位群体。最终将一个主效QTL定位于chr2-2的引物CHR2-9和CHR2-10之间,该QTL对粒长,粒宽,粒厚及千粒重均有很大的效应(图4),我们命名为qGS2。表3大粒基因关联分析引物在CHR2-9和CHR2-10之间根据测序结果进一步设计引物(表4),扩大定位群体BC4F2至5210株,最终将qGS2基因定位在CHR2-30和CHR2-21之间的39.5kb区间内。表4GS2基因精细定位引物对水稻互作基因的定位:两个水稻叶片长茸毛发生相关基因的精细定位菲B10是一份叶片带有长茸毛的品种,而cypress是一份光叶水稻,两者的杂交F1表现为长茸毛性状。以cypress为轮回亲本,并在F1中选取长茸毛植株进行多代回交,获得BC4F1。对31株BC4F1的长茸毛性状进行调查,其中8株为长茸毛,其他23株为非长茸毛,分离比接近3:1,推测长茸毛为两个基因控制。从BC4F1分别选取一株带长茸毛单株(HL)及一株光叶(GL)进行基因组重测序。通过测序,GL获得高质量的reads51552068条,7499073075bp,平均覆盖率大于19.55倍,覆盖了水稻基因组的94.7%的区段,HL获得高质量的reads70293248条,10228526261bp,平均覆盖率大于26.72倍,覆盖了水稻基因组的95.07%的区段。与参考基因组序列对比后GL获得多态1695933个,HL获得多态1880781个,其中我们选取depth>=5,变异频率在0.2至0.8之间的为可信杂合位点,depth>=5,变异频率为1的为可信的纯合位点进行下一步分析。对比两组多态,我们获得差异多态HL179477个,GL为80943个,统计500kb区间内两份材料差异多态的分布(表5),并进行比对,如图5所示,我们分别在第2,3,5,6号染色体上获得了一个差异区段。利用测序结果设计这4个区间内的indel标记(表6),对31株BC4F1进行关联分析。结果表明chr5和chr6两个区段与长茸毛性状共分离,当这两个位点都为杂合时表现为长茸毛,两个位点都是纯合时表现为光叶,仅有一个位点杂合时表现为无茸毛,并推测这两个基因间的互作为累加效应(additiveeffect)(表7)。将两个位点分别命名为LH5和LH6。利用测序单株HL的后代276株BC4F2群体,并根据测序结果设计引物进行定位分析(表8),我们将LH5定位在引物LH5-11与LH5-7之间的228kb范围内,LH6定位在引物LH6-4与LH6-6之间406kb范围内。表5多态统计表表6水稻叶片表面长茸毛相候选位点关联分析引物表7利用31株BC4F1对差异位点进行连锁分析表8水稻叶片表面长茸毛相关基因M5和M6的精细定位对定位区间内的基因进行分析,我们认为LH5即为前人克隆的光叶基因WOX3,而LH6为一新基因,选取BC4F2中LH5为WOX3,LH6为杂合的单株后代2300株对LH6进行精细定位。首先对2300株后代在引物M6-4和引物M6-6之间的重组单株进行筛选,共筛选到27株,在该区间内设计indel引物5对,将该基因最终定位在58.8kb范围内(图6)。讨论重测序技术因其准确、高效的特点目前已被广泛应用于基因定位中,包括对突变基因、质量性状基因的克隆,对QTLs的定位也有报道。本发明则提供了一种新的利用重测序来定位克隆QTLs的策略。与传统图位克隆相比,通过重测序可获得足够多的分子标记用于定位,在选择亲本时只需考虑目标性状差异而无需考虑亲本间的遗传多态性,亲本的选择余地更大。例如在本实验中我们选用的材料为两份籼稻材料,在利用SSR标记对两份材料间的多态进行筛选时多态率低于10%,一条染色体上仅能找到1-3对有多态的SSR标记,这样的多态分布显然不符合定位的要求。通过重测序,我们仅在目标基因附近1000kb(chr2:30000000-31000000)内就找到INDEL标记94个,SNP标记822个,因此有足够丰富的多态以供选择。传统的QTL定位时,往往需要先用初级群体对QTL进行初步定位,然后结合分子标记辅助选择构建近等基因系等群体进行精细定位,工作量非常巨大。而本研究先直接构建高世代回交材料,先对遗传背景进行了同一化处理;利用重测序快速确定候选区域,仅需利用差异区间内的少数几对引物及相对较小的一个定位群体即能将获得QTLs精细定位。同时在对基因进行定位分析时,我们仅选取了差异区段内的12对引物对76株BC4F2进行连锁分析,即获得了目标QTLs,大大减少了工作量。在将重测序技术引入到QTLs定位的QTL-seq技术中,其原理是对F2或重组自交系中性状分离的20-50个单株混样进行重测序,快速的获得QTLs。通过该技术能快速获得QTLs,但要克隆到该目标性状,仍需要进一步构建分离群体,尤其是复杂性状的克隆,通常还是需要获得NearlyIsogenicLines(NILs)来克隆该基因,并进一步利用NILs来验证其功能。与QTL-seq相比在新的策略里是先获得高代回交材料,从回交F2代中,利用测序获得的标记对遗传背景进行筛选,我们能很快获得NILs,这样就在定位到QTLs的同时也获得了克隆目标基因所需要的NILs,因此在通过重测序后很快就能有效的克隆到目标基因。同时在对重测序数据进行分析时,我们也采用了与QTL-seq技术不同的方法。在QTL-seq中至少要已知其中一方亲本的序列,将其作为参照基因组与两份待测材料进行比对,分析SNP-index即统计材料与参照序列不同的SNP出现的频率来确定QTLs。而在本发明中所用材料无需是已知序列材料,也无需对亲本进行测序分析,测序的结果只需与已知的水稻序列比较,如网上提供的粳稻日本晴序列,籼稻9311序列等。通过分析与参考序列多态位点的杂合属性来确定QTLs候选区域,分析方法更简便直接,材料要求更低。相比于以往的技术,该技术能更有效的对目标QTLs进行克隆,而不是仅仅停留在分离定位这一阶段。尤其在育种过程中,我们通常会将一些通过反复回交的方式将某一优良性状导入到其他品种当中,以进一步改良该品种。在此过程中应用新的策略不仅能对一些基因进行克隆,也能使这些基因更快的应用于育种中。同时我们也预想如果回交的群体足够大,我们也能将与目标性状相关的基因逐一克隆到。以上所述的实施例只是本发明的一种较佳的方案,并非对本发明作任何形式上的限制,在不超出权利要求所记载的技术方案的前提下还有其它的变体及改型。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1