一种缩酚酸环醚化合物及其制备方法和应用与流程
本发明属于植物有效成分提取
技术领域:
,具体涉及一种缩酚酸环醚化合物及其制备方法和应用。
背景技术:
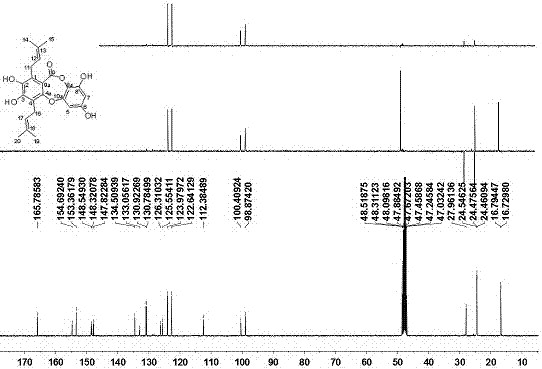
:藤黄科(garcinial.)植物全球约450种,产亚洲、非洲南部及波利尼西亚西部,我国有21种,分布于广东、广西、云南等南部省区。藤黄科植物还是天然呫吨酮(xanthone)类成分的主要资源之一,富含异戊烯基取代的呫吨酮(xanthones),这类成分结构新颖多样,且具有广泛的药理活性,尤以藤黄酸(gambogicacid)最具代表性,具有广谱强效的抗肿瘤活性,是近年来抗肿瘤天然产物的研究热点之一,我国学者正在开发其注射液,为抗肿瘤一类新药。除呫吨酮(xanthone)类外,缩酚酸环醚化合物(depsidone)、苯甲酮类(benzophenones)、双黄酮类(bioflavonoids)和联苯类(biphenyls)等化合物也是本科植物的特征性成分,也具有多种生物活性。为了更有效地利用我国藤黄科植物资源,从中寻找具有开发前景的活性成分,我们选择对藤黄科植物开展较系统的活性成分研究工作。技术实现要素:本发明的第一目的是提供一种缩酚酸环醚化合物;第二目的在于提供所述缩酚酸环醚化合物的制备方法;第三目的在于提供所述缩酚酸环醚化合物在制备抗轮状病毒药物中的应用。本发明的第一目的是这样实现的,所述的缩酚酸环醚化合物是以干燥藤黄科乔木枝条、叶或果实为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离得到的,该化合物分子式为c23h24o7,该化合物命名为multidepsidonea,具有下述结构式:本发明的第二目的是这样实现的,所述的缩酚酸环醚化合物的制备方法,是以干燥藤黄科乔木枝条、叶或果实为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离获得,具体为:a、浸膏提取:将藤黄科科乔木枝条、叶或果实粗粉碎到20~40目,用有机溶剂超声提取2~4次,每次30~60min,提取液合并;提取液过滤,减压浓缩提取液至1/4~1/2体积时,静置,滤除沉淀物,浓缩成浸膏a;b、有机溶剂萃取:浸膏a中加入重量比1~2倍量的水,用与水等体积的有机溶剂萃取3~5次,合并有机溶剂萃取相,减压浓缩成浸膏b;c、硅胶柱层析:将浸膏b用重量比1.5~3倍量的有机溶剂溶解,然后用浸膏重0.8~1.2倍的200~300目硅胶拌样,然后上硅胶柱层析,装柱硅胶为200~300目,用量为浸膏b重量6~8倍量;用体积比为1:0~0:1的混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;d、反相柱层析:将以4:1配比的有机溶剂进行洗脱得到的洗脱液上反相柱层析,反相柱是用c-18、c-8或ods等反相材料装柱;用体积含量为40~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;e、高效液相色谱分离:将以体积含量55~80%甲醇水溶液洗脱得到的洗脱液经高效液相色谱分离纯化,即得所述的缩酚酸环醚化合物multidepsidonea;f、e步骤所述的高效液相色谱分离纯化是以50~70%的甲醇为流动相,流速10~14ml/min,21.2×250mm,5um的反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样45~60ul,收集16~34min的色谱峰,多次累加后蒸干。即得所述的缩酚酸环醚化合物multidepsidonea。本发明缩酚酸环醚化合物是首次被分离出来的,通过核磁共振和其他波谱技术测定方法确定为缩酚酸环醚化合物,并表征其具体结构为:化合物multidepsidonea,为橙黄色胶状物;紫外光谱(溶剂为甲醇),λmax(logε):274(3.29),205(3.98)nm;红外光谱(溴化钾压片)υmax3425,2968,2925,1714,1615,1595,1508,1484,1266,1144,1080,996,838cm–1;hresims显示本发明化合物准分子离子峰m/z412.1514[m]+(计算值为412.1522),结合13c和1hnmr谱(图1和图2,碳谱氢谱数据归科见表1)给出其分子式为c23h24o7。1hnmr(cd3od,400mhz)和13cnmr(cd3od,100mhz)数据,见表1。hresims显示其准分子离子峰为准分子离子峰m/z412.1514[m]+(计算值为412.1522),结合13cnmr谱确定分子式为c23h24o7,不饱和度为12。红外光谱显示了羟基(3425cm-1)、内酯羰基(1714cm-1)的吸收峰。在13c-nmr中,c-9(δc165.8)也证实了内酯羰基的存在。该化合物的1h-和13c-nmr数据归科,显示其含有23个碳,其中包括4个甲基、1个羰基、2个亚甲基、4个烯次甲基、12个季碳。根据核磁共振数据以及两组典型的异戊烯基信号[δh6.41(1h,d,j=2.7hz,h-5),6.18(1h,d,j=2.7hz,h-7),4.96–4.98(1h,m,h-12),4.92–4.95(1h,m,h-17),3.37(2h,d,j=6.0hz,h-11),3.28(2h,d,j=6.6hz,h-16),1.68(3h,s,h-14),1.62(3h,s,h-20),1.61(3h,s,h-15),和1.60(3h,s,h-19);δc28.0(c-11),124.0(c-12),130.9(c-13),16.8(c-14),24.5(c-15),24.5(c-16),122.6(c-17),130.8(c-18),16.7(c-19),24.5(c-20)]可初步推测该化合物为含有两个异戊烯基的缩酚酸环醚类化合物。根据芳香质子信号δh6.41与c-6、c-7、c-10a、c-8a以及δh6.18与c-5,c-6,c-8,c-8a呈现hmbc相关,可确定h-5(δh6.41)和h-7(δh6.18)为苯环上间位氢,进一步能确定其具有一个4取代的芳香环。h-11与c-1、c-2、c-9a呈现hmbc相关,以及h-12与c-1呈现hmbc相关,证实c-1上连有一个异戊烯基。根据h-16和h-17与c-4(δc125.6)呈现hmbc相关,可以证实另一个异戊烯基与c-4相连。根据c-2(δc134.5),c-3(δc148.3),c-6(δc153.4),c-8(δc154.7)为季碳以及其分子式为c23h24o7,可以确定4个羟基分别取代在c-2,c-3,c-6和c-8。至此,该化合物的结构得到确定,将其命名为multidepsidonea。本发明的第三目的是这样实现的,所述的缩酚酸环醚化合物在制备抗轮状病毒药物中的应用。经抗轮状病毒活性实验,选用病毒唑作为对照,multidepsidonea对轮状病毒的cc50和ec50值分别为290.3和19.2μmol/l,,其具有较好的抗轮状病毒活性。本发明化合物结构简单活性好,可作为抗轮状病毒药物的先导性化合物,有良好的应用前景。附图说明图1为化合物multidepsidonea的核磁共振碳谱(13cnmr);图2为化合物multidepsidonea的核磁共振氢谱(1hnmr);图3为化合物multidepsidonea的主要hmbc(→)相关。具体实施方式下面结合附图对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或改进,均落入本发明的保护范围。本发明所述的缩酚酸环醚化合物是以干燥藤黄科乔木枝条、叶或果实为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离得到的,该化合物分子式为c23h24o7,命名为multidepsidonea,具有下述结构式:本发明的第二目的是这样实现的,所述的缩酚酸环醚化合物的制备方法,是以干燥藤黄科乔木枝条、叶或果实为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离获得,具体为:a、浸膏提取:将藤黄科乔木枝条、叶或果实粗粉碎到20~40目,用有机溶剂超声提取2~4次,每次30~60min,提取液合并;提取液过滤,减压浓缩提取液至1/4~1/2体积时,静置,滤除沉淀物,浓缩成浸膏a;b、有机溶剂萃取:浸膏a中加入重量比1~2倍量的水,用与水等体积的有机溶剂萃取3~5次,合并有机溶剂萃取相,减压浓缩成浸膏b;c、硅胶柱层析:将浸膏b用重量比1.5~3倍量的有机溶剂溶解,然后用浸膏重0.8~1.2倍的200~300目硅胶拌样,然后上硅胶柱层析,装柱硅胶为200~300目,用量为浸膏b重量6~8倍量;用体积比为1:0~0:1的混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;d、反相柱层析:将以4:1配比的有机溶剂进行洗脱得到的洗脱液上反相柱层析,反相柱是用c-18、c-8或ods等反相材料装柱;用体积含量为40~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;e、高效液相色谱分离:将以体积含量55~80%甲醇水溶液洗脱得到的洗脱液经高效液相色谱分离纯化,即得所述的缩酚酸环醚化合物multidepsidonea;f、e步骤所述的高效液相色谱分离纯化是以50~70%的甲醇为流动相,流速10~14ml/min,21.2×250mm,5um的反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样45~60ul,收集16~34min的色谱峰,多次累加后蒸干。本发明所述的缩酚酸环醚化合物在制备抗轮状病毒药物中的应用。本发明所述的藤黄科植物不受地区和品种限制,均可以实现本发明。实施例1取干燥的藤黄科乔木枝条、叶和/或果实5.6kg,粗粉碎至20目,用70%的丙酮超声提取4次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/4;静置,滤除沉淀物,浓缩成660g浸膏a;在浸膏a中加入660g水,用与水等体积的氯仿萃取5次,合并萃取相,减压浓缩成244g浸膏b;用200目硅胶1700g装柱,在浸膏b中加入600g的丙酮溶解,然后加入100目硅胶290g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、7:3、3:2、1:1、1:2、0:1的氯仿-甲醇混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到9个部分,体积比7:3的氯仿-甲醇混合有机溶剂的洗脱液c为57g;用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以60%的甲醇为流动相,流速8ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ul,收集21min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例2取干燥的藤黄科乔木枝条、叶和/或果实3.2kg,粗粉碎至20目,用100%的乙醇超声提取2次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/3;静置,滤除沉淀物,浓缩成360g浸膏a;在浸膏a中加入360g的水,用与水等体积的氯仿萃取3次,合并萃取相,减压浓缩成120g浸膏b;用160目硅胶960g装柱,在浸膏b中加入240g的丙酮溶解,然后加入80目硅胶120g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、3:2、1:1、1:2、0:1的正己烷-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的正己烷-丙酮混合有机溶剂的洗脱液c为46g;用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以68%的甲醇为流动相,流速14ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ul,收集23min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例3取干燥的藤黄科乔木枝条、叶和/或果实6.5kg,粗粉碎至30目,用80%的甲醇超声提取4次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/2;静置,滤除沉淀物,浓缩成675g浸膏a;在浸膏a中加入700g的水,用与水等体积的乙醚萃取4次,合并萃取相,减压浓缩成342g浸膏b;用180目硅胶2800g装柱,在浸膏b中加入930g的丙酮溶解,然后加入90目硅胶360g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、7:3、3:2、1:1、1:2、0:1的氯仿-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的氯仿-丙酮混合有机溶剂的洗脱液c为45g;用反相材料ods装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以55%的甲醇为流动相,流速12ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ul,收集20min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例4取干燥的藤黄科乔木枝条、叶和/或果实5.9kg,粗粉碎至40目,用90%的乙醇提取3次,提取液合并;提取液过滤,减压浓缩至体积的1/4;静置,滤除沉淀物,浓缩成810g浸膏a;在浸膏a中加入880g的水,用与水等体积的石油醚萃取4次,合并萃取相,减压浓缩成265g浸膏b;用160目硅胶1450g装柱,在浸膏b中加入290g的丙酮溶解,然后加入80目硅胶265g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、7:3、3:2、1:1、1:2、0:1的石油醚-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的石油醚-丙酮混合有机溶剂的洗脱液c为52g;用反相材料c-8装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以70%的甲醇为流动相,流速10ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,收集18min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例5取干燥的藤黄科乔木枝条、叶和/或果实5.6kg,粗粉碎至20目,用70%的甲醇超声提取4次,每次35min,提取液合并;提取液过滤,减压浓缩至体积的1/2;静置,滤除沉淀物,浓缩成700g浸膏a;在浸膏a中加入1400g的水,用与水等体积的苯萃取5次,合并萃取相,减压浓缩成310g浸膏b;用200目硅胶1860g装柱,在浸膏b中加入420g的丙酮溶解,然后加入100目硅胶310g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、3:2、1:1、1:2、0:1的石油醚-乙酸乙酯混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的石油醚-乙酸乙酯混合有机溶剂的洗脱液c为56g;用反相材料ods装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以50%的甲醇为流动相,流速12ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,收集26min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例6取干燥的藤黄科乔木枝条、叶和/或果实11kg,粗粉碎至20目,用100%的乙醇超声提取2次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/3;静置,滤除沉淀物,浓缩成1200g浸膏a;在浸膏a中加入1200g的水,用与水等体积的氯仿萃取3次,合并萃取相,减压浓缩成400g浸膏b;用160目硅胶3200g装柱,在浸膏b中加入400g的丙酮溶解,然后加入80目硅胶400g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、3:2、1:1、1:2、0:1的正己烷-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的氯仿-丙酮混合有机溶剂的洗脱液c为110g;用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以68%的甲醇为流动相,流速14ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ul,收集22min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例7取干燥的藤黄科乔木枝条、叶和/或果实1kg,粗粉碎至30目,用80%的甲醇超声提取4次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/2;静置,滤除沉淀物,浓缩成110g浸膏a;在浸膏a中加入120g的水,用与水等体积的乙醚萃取4次,合并萃取相,减压浓缩成52g浸膏b;用180目硅胶430g装柱,在浸膏b中加入140g的丙酮溶解,然后加入90目硅胶55g拌样,拌样后上柱;用体积比分别为1:0、20:1、9:1、8:2、7:3、3:2、1:1、1:2、0:1的氯仿-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比9:1的氯仿-丙酮混合有机溶剂的洗脱液c为6g;用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量50~70%甲醇水溶液洗脱得到的洗脱液,再以55%的甲醇为流动相,流速12ml/min,21.2mm×25cm,5um的zorbaxprephtgf反相制备柱为固定相,紫外检测器检测波长为254nm,每次进样50ul,收集20min的色谱峰,多次累加后蒸干,即得所述的缩酚酸环醚化合物multidepsidonea。实施例8取实施例1制备的化合物multidepsidonea,为橙黄色胶状物;测定方法为:用核磁共振,结合其它波谱技术鉴定结构。(1)紫外光谱(溶剂为甲醇),λmax(logε):274(3.29),205(3.98)nm;(2)红外光谱(溴化钾压片)υmax3425,2968,2925,1714,1615,1595,1508,1484,1266,1144,1080,996,838cm–1;(3)hresims显示本发明化合物准分子离子峰为m/z412.1514[m]+(计算值为412.1522),结合13c和1hnmr谱(图1和图2,碳谱氢谱数据归科见表1)给出其分子式为c23h24o7。1hnmr(400mhz,cd3od)和13cnmr(100mhz,cd3od)数据,见表1。hresims显示其准分子离子峰为m/z412.1514[m]+(计算值为412.1522),结合13cnmr谱确定分子式为c23h24o7,不饱和度为12。红外光谱显示了羟基(3425cm-1)、内酯羰基(1714cm-1)的吸收峰。在13c-nmr中,c-9(δc165.8)也证实了内酯羰基的存在。该化合物的1h-和13c-nmr数据归科,显示其含有23个碳,其中包括4个甲基、1个羰基、2个亚甲基、4个烯次甲基、12个季碳。根据核磁共振数据以及两组典型的异戊烯基信号[δh6.41(1h,d,j=2.7hz,h-5),6.18(1h,d,j=2.7hz,h-7),4.96–4.98(1h,m,h-12),4.92–4.95(1h,m,h-17),3.37(2h,d,j=6.0hz,h-11),3.28(2h,d,j=6.6hz,h-16),1.68(3h,s,h-14),1.62(3h,s,h-20),1.61(3h,s,h-15),和1.60(3h,s,h-19);δc28.0(c-11),124.0(c-12),130.9(c-13),16.8(c-14),24.5(c-15),24.5(c-16),122.6(c-17),130.8(c-18),16.7(c-19),24.5(c-20)]可初步推测该化合物为含有两个异戊烯基的缩酚酸环醚类化合物。根据芳香质子信号δh6.41与c-6、c-7、c-10a、c-8a以及δh6.18与c-5,c-6,c-8,c-8a呈现hmbc相关,可确定h-5(δh6.41)和h-7(δh6.18)为苯环上间位氢,进一步能确定其具有一个4取代的芳香环。h-11与c-1、c-2、c-9a呈现hmbc相关,以及h-12与c-1呈现hmbc相关,证实c-1上连有一个异戊烯基。根据h-16和h-17与c-4(δc125.6)呈现hmbc相关,可以证实另一个异戊烯基与c-4相连。根据c-2(δc134.5),c-3(δc148.3),c-6(δc153.4),c-8(δc154.7)为季碳以及其分子式为c23h24o7,可以确定4个羟基分别取代在c-2,c-3,c-6和c-8。至此,该化合物的结构得到确定,将其命名为multidepsidonea。实施例9取实施例2制备的化合物,为橙黄色胶状物;按实施例8中的方法进行结构测定,结果为:其结构同实施例8,分子式为c23h24o7。确认实施例2制备的化合物为所述的缩酚酸环醚化合物multidepsidonea。实施例10取实施例3制备的化合物,为橙黄色胶状物;按实施例8中的方法进行结构测定,结果为:其结构同实施例8,分子式为c23h24o7。确认实施例3制备的化合物为所述的缩酚酸环醚化合物multidepsidonea。表1化合物的1h和13cnmr数据(400/100mhz,cd3od)noδcδh(m,j,hz)noδcδh(m,j,hz)1133.0s9a112.4s2134.5s1128.0t3.37(d,6.0)3148.3s12124.0d4.96–4.98(m)4125.6s13130.9s4a148.6s1416.8q1.68(s)598.9d6.41(d,2.7)1524.5q1.61(s)10a147.8s1624.5t3.28(d,6.6)6153.4s17122.6d4.92–4.95(m)7100.4d6.18(d,2.7)18130.8s8154.7s1916.7q1.60(s)8a126.3s2024.5q1.62(s)9165.8s实施例11取实施例4制备的化合物,为橙黄色胶状物;按实施例8中的方法进行结构测定,结果为:其结构同实施例8,分子式为c23h24o7。确认实施例4制备的化合物为所述的缩酚酸环醚化合物multidepsidonea。实施例12取实施例5制备的化合物,为橙黄色胶状物;按实施例8中的方法进行结构测定,结果为:其结构同实施例8,分子式为c23h24o7。确认实施例5制备的化合物为所述的缩酚酸环醚化合物multidepsidonea。实施例11取实施例1~7所制备的任一缩酚酸环醚化合物进行抗轮状病毒活性检测试验,试验情况如下:细胞株:恒河猴肾细胞系(ma-104)。实验设计:ma-104细胞与不同浓度化合物温育72小时,每株细胞的实验均重复3次,用3次实验的结果进行数据处理,采用改良mtt法评价化合物对细胞增殖的抑制程度,计算抑制率,根据抑制率采用logit方法计算ic50,比较化合物的体外抗病毒活性。ec50即指半数有效浓度,是指引起50%试验动物产生某一特定反应,或是某反应指标被抑制一半时的浓度。cc50即指半数细胞毒性浓度,是指对半数细胞产生毒性作用所需浓度。在本实验中,指致50%细胞死亡所需的药物浓度。化合物细胞毒性测定化合物用二甲基亚砜(dimethylsulfoxide,dmso)溶解,于微波消毒l0min,用mem配成lmg/ml的母液备用,mem溶液稀释成所需浓度。96孔细胞培养板,加lxl05/ml浓度的mal04细胞悬液,100ul/孔,37℃、5%co2孵箱培养24h,在生长良好的单层细胞上分别加入浓度分别为lmg/ml、0.2mg/ml、40ug/ml、8ug/ml、1.25ug/ml的化合物;100ul/孔,每个浓度设3个复孔,同时设正常细胞对照。置于37℃,5%co2孵箱继续培养24h后,mtt法检测细胞存活率。化合物对病毒感染预防作用以细胞浓度为104/ml,每孔l00ul接种细胞于96孔板中,培养24小时,见细胞长成单层并生长状况良好,用浓度分别为100ug/ml、75ug/ml、50ug/ml、25ug/ml、lug/ml的化合物在37℃孵箱预先作用细胞l.5h,pbs洗涤后以100tcid50/ml的轮状病毒每孔100ul吸附lh后弃去,加mem培养基100ul/孔维持,置37c、5%co2孵箱,每日观察细胞病变情况。48h后以mtt法检测病毒抑制率。化合物对病毒感染治疗作用以细胞浓度为104/ml,每孔l00ul接种细胞于96孔板中,培养24小时,见细胞长成单层并生长状况良好,先用以100tcid50/ml的轮状病毒每孔100ul吸附lh后弃去,然后加入上述不同浓度的化合物,100ul/孔,同上法培养与检测。各组实验均设病毒对照组(c组)和正常细胞对照组(n组)。细胞存活率测定采用mtt法,在培养48h的细胞中加入5mg/ml甲基噻唑基四唑(methylthiazolyltetrazolium,mtt)20ul,继续培养3-4h,弃上清,加入dmso每孔100ul,振荡使孔内结晶完全溶解后立即在490lun波长下测定吸光度a值。细胞存活率=药物组平均a值/细胞对照组a值x100%病毒抑制率=[实验组平均a值一病毒对照组平均a值]/[细胞对照组平均a值一病毒对照组平均a值]x100%治疗指数(ti)=半数毒性浓度(cc50)/半数抑制浓度(ic50)(e)实验结果实验结果表明:经抗轮状病毒活性实验,选用病毒唑作为对照,multidepsidonea对轮状病毒的cc50和ec50值分别为290.3和19.2μmol/l,其具有较好的抗轮状病毒活性。表2化合物的抗轮状病毒活性no.cc50(µm)ec50(µm)timultidepsidonea290.319.215.12ribavirin263.213.319.8a所有数据均表示为平均值±sd(标准差);n=3si:选择指数,cc50/ec50。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1