大黄鱼干扰素d基因启动子序列及其应用的制作方法
本发明涉及干扰素IFNd基因启动子,尤其是涉及大黄鱼干扰素IFNd基因启动子序列及其应用。
背景技术:
干扰素(Interferon,IFN)是一类能诱导脊椎动物细胞抗病毒状态,并在抗病毒防御中起重要作用的诱导性多基因家族细胞因子(1.Robertsen B.The interferon system of teleost fish[J].Fish Shellfish Immunol,2006,20(2):172-191)。硬骨鱼类作为低等脊椎动物也拥有相对完善的干扰素系统。鱼类IFNs分为I型IFNs和II型IFNs两大类,其中I型IFNs因其专一性地协调宿主抗病毒免疫而受到更多的关注(2.Zhang YB,Gui JF.Molecular regulation of interferon antiviral response in fish[J].Dev Comp Immunol,2012,38(2):193-202.)。根据成熟肽中半胱氨酸的数量及分布,鱼类I型IFNs分为group I IFN(成熟肽含2个半胱氨酸)和group II IFN(成熟肽含4个半胱氨酸)。Group I IFN包括IFNa、IFNd和IFNe 3个亚组,group II IFN包括IFNb、IFNc和IFNf3个亚组(3.Zou J,Gorgoglione B,Taylor NG,et al.Salmonids have an extraordinary complex type I IFN system:characterization of the IFN locus in rainbow trout oncorhynchus mykiss reveals two novel IFN subgroups[J].J Immunol,2014,193(5):2273-2286)。基因的表达调控已成为分子生物学研究领域的热点,启动子是基因表达调控的重要元件。鉴于鱼类I型IFNs在抗病毒免疫及病毒病防治中的重要性,研究其基因表达调控机制将为通过调节IFN的表达来防治鱼类病毒病提供新的思路。对鱼类I型IFNs基因5′侧翼调控区序列的分析表明,鱼类I型IFNs基因启动子存在多个转录因子结合位点,如IRF3、IRF7、NF-κB和ISRE等,并且具有复杂的表达调控机制。
本申请人在前期通过全基因组序列分析,发现了1条编码大黄鱼I型IFN的基因序列(4.Ao JQ,Mu YN,Xiang LX,et al.Genome sequencing of the perciform fish Larimichthys crocea provides insights into stress adaptation[J].Plos Genet,2015,11(4):e1005118)。同源比对及进化分析表明该I型IFN为IFNd,属于鱼类group I IFNs。由于启动子是决定基因表达及其调控的关键因素,为研究大黄鱼IFNd基因的表达调控机制,从其基因组进一步克隆了大黄鱼IFNd基因的5′侧翼启动子序列,经报告基因检测实验证明了该大黄鱼IFNd基因启动子具有较强的启动子活性。该大黄鱼IFNd基因启动子的克隆及其启动子活性的验证,在理论上将为研究大黄鱼IFNd基因的表达调控机理提供良好的实验系统,在应用方面为利用该强启动子构建表达载体高效表达外源基因或将该启动子应用于转基因鱼构建创造了条件,具有重要的理论和实际意义。
技术实现要素:
本发明的目的之一在于提供大黄鱼IFNd基因启动子的序列。
本发明的目的之二在于提供一种大黄鱼IFNd基因启动子的活性分析方法。
本发明的目的之三在于提供大黄鱼IFNd基因启动子的应用。
所述大黄鱼IFNd基因启动子的序列为:
所述大黄鱼IFNd基因启动子的活性分析方法如下:
1)采用Promega公司双荧光素酶报告基因检测系统定量分析所述大黄鱼IFNd基因启动子的活性以及人工合成双链RNA poly(I:C)、大肠杆菌脂多糖LPS、枯草芽孢杆菌肽聚糖PGN和嗜水气单胞菌基因组DNA刺激对其活性的影响;
2)将所述大黄鱼IFNd基因启动子区片段插入Promega公司荧光素酶报告基因载体pGL3-Basic中,使萤火虫荧光素酶基因位于该启动子的控制下,构建得到重组载体pGL3-IFNdP;用重组载体pGL3-IFNdP和海肾荧光素酶报告基因载体pRL-TK共同转染EPC细胞,48h后收集转染细胞;利用双荧光素酶报告基因检测系统分别检测萤火虫荧光素酶和海肾荧光素酶的酶活性值,通过计算二者酶活性值的比值得出转染细胞中荧光素酶的相对活性;
3)以空载体pGL3-Basic和海肾荧光素酶对照报告基因载体pRL-TK共同转染EPC细胞的荧光素酶相对活性作为对照,计算出该启动子的相对活性,再用poly(I:C)、脂多糖LPS、肽聚糖PGN及细菌基因组DNA刺激pGL3-IFNdP转染的EPC细胞,通过分析各处理组荧光素酶相对活性的变化,来确定大黄鱼IFNd基因启动子的活性是否受刺激物诱导的影响。
所述大黄鱼IFNd基因启动子的应用如下:
1)所述大黄鱼IFNd基因启动子的克隆及其启动子活性的鉴定,在构建大黄鱼IFNd基因的表达调控机理实验系统中应用;
2)所述大黄鱼IFNd基因启动子可在构建真核表达载体在鱼类细胞或哺乳动物细胞中高效表达外源基因中应用;
3)所述大黄鱼IFNd基因启动子在构建转基因鱼中应用。
本发明的突出优点和技术效果如下:
本发明采用基因组步移的方法克隆了大黄鱼IFNd基因启动子,提供了大黄鱼IFNd基因启动子序列;经报告基因分析实验证明了该大黄鱼IFNd基因启动子可被poly(I:C)、LPS、PGN及细菌基因组DNA诱导激活。因此,该大黄鱼IFNd基因启动子的克隆及其强启动子活性的验证,在理论上将为研究大黄鱼IFNd基因的表达调控机理提供良好的实验系统,在应用方面为利用该启动子构建表达载体高效表达外源基因或将该启动子应用于转基因鱼构建创造了条件,具有重要的理论和实际意义。
附图说明
图1为采用双荧光素酶报告基因检测系统定量分析大黄鱼IFNd基因启动子的活性。横坐标pGL3表示空载体pGL3-Basic转染EPC细胞的荧光素酶相对活性(作为对照);pGL3-IFNdP为重组载体pGL3-IFNdP转染EPC细胞的荧光素酶相对活性。如图1所示,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的5.6倍,说明了大黄鱼IFNd基因启动子可以较好地启动荧光素酶报告基因的转录。每个实验设三次重复,每次重复设三个平行;误差靶代表平均值的标准差。
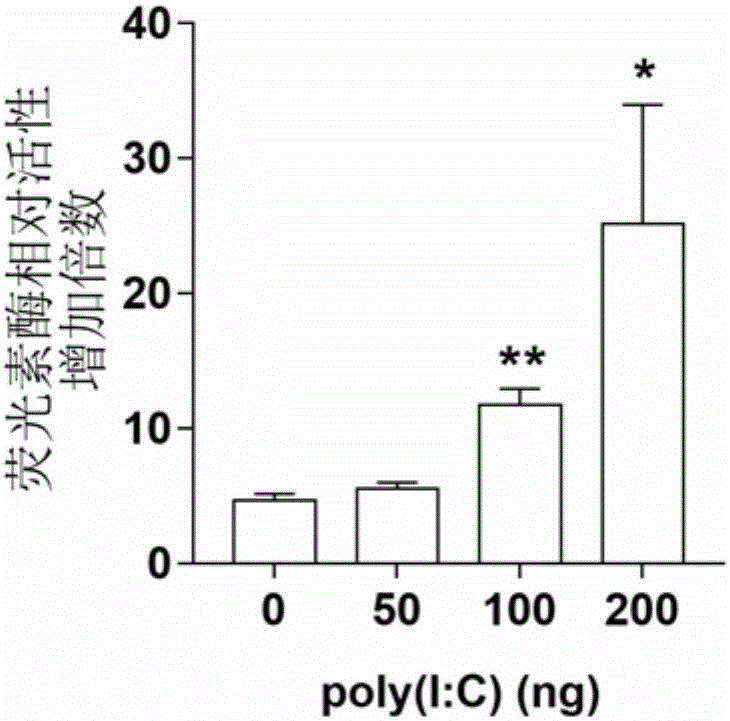
图2为在不同剂量poly(I:C)刺激条件下大黄鱼IFNd基因启动子的活性变化。横坐标分别表示在0ng、50ng、100ng、200ng poly(I:C)刺激条件下重组载体pGL3-IFNdP转染EPC细胞的荧光素酶相对活性。纵坐标表示在转染相同剂量poly(I:C)条件下,重组载体pGL3-IFNdP转染EPC细胞中荧光素酶相对活性相对于空载体pGL3-Basic转染EPC细胞的荧光素酶相对活性(定义为1)的增加倍数。如图2所示,在poly(I:C)剂量为0ng、50ng、100ng及200ng时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.7倍、5.6倍、11.8倍及25.1倍。说明了该大黄鱼IFNd基因启动子可被poly(I:C)诱导激活。每个实验设三次重复,每次重复设三个平行。误差靶代表平均值的标准差。利用双尾T检验fa计算处理组与对照组的显著性差异,*p<0.05,**p<0.01.
图3为在不同浓度LPS刺激条件下大黄鱼IFNd基因启动子的活性变化。横坐标分别表示在0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL LPS刺激条件下重组载体pGL3-IFNdP转染EPC细胞的荧光素酶相对活性。纵坐标表示在相同浓度LPS刺激条件下,重组载体pGL3-IFNdP转染EPC细胞中荧光素酶相对活性相对于空载体pGL3-Basic转染EPC细胞的荧光素酶相对活性(定义为1)的增加倍数。如图3所示,在LPS浓度为0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.6倍、5.8倍、7.2倍、7.4倍及7.4倍。该大黄鱼IFNd基因启动子可被LPS诱导激活。*p<0.05,**p<0.01.
图4为在不同浓度PGN刺激条件下大黄鱼IFNd基因启动子的活性变化。横坐标分别表示在0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL LPS刺激条件下重组载体pGL3-IFNdP转染EPC细胞的荧光素酶相对活性。纵坐标表示在相同浓度PGN刺激条件下,重组载体pGL3-IFNdP转染EPC细胞中荧光素酶相对活性相对于空载体pGL3-Basic转染EPC细胞的荧光素酶相对活性(定义为1)的增加倍数。如图4所示,在PGN浓度为0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.6倍、6.1倍、7.5倍、6.6倍及5.6倍。该大黄鱼IFNd基因启动子可被PGN诱导激活。*p<0.05,**p<0.01.
图5为在不同剂量细菌基因组DNA刺激条件下大黄鱼IFNd基因启动子的活性变化。横坐标分别表示在0ng、10ng、20ng、50ng、100ng细菌基因组DNA刺激条件下重组载体pGL3-IFNdP转染EPC细胞的荧光素酶相对活性。纵坐标表示在转染相同剂量细菌基因组DNA条件下,重组载体pGL3-IFNdP转染EPC细胞中荧光素酶相对活性相对于空载体pGL3-Basic转染EPC细胞的荧光素酶相对活性(定义为1)的增加倍数。如图5所示,在细菌基因组DNA剂量为0ng、10ng、20ng、50ng、100ng时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.4倍、4.5倍、6.9倍、10.2倍及10.7倍。该大黄鱼IFNd基因启动子可被细菌基因组DNA诱导激活。*p<0.05,**p<0.01.
具体实施方式
以下实施例将结合附图对本发明作进一步说明。
1.大黄鱼IFNd基因启动子的克隆
1.1采用Clontech公司的GenomeWalker Universal Kit试剂盒分别构建4种限制性内切酶的大黄鱼基因组单酶切文库,所用的4种DNA核酸内切酶为Dra I、EcoR V、PvμII和StμI。具体操作为:
1.1.1基因组纯度分析。
①取1μL大黄鱼基因组DNA在0.6%的琼脂糖凝胶上电泳。
②用核酸内切酶Dra I试切。体系为:
1.1.2基因组酶切消化。
①标记5个1.5mL的离心管DL1、DL2、DL3、DL4和一个阳性对照。
②各内切酶酶切体系组分包括:
③低速涡旋5~10s并瞬时离心,37℃过夜消化。
1.1.3基因组DNA纯化。
①在上述每一个反应管中加入等体积的苯酚。
②低速涡旋5~10s并瞬时离心。
③把上层水相转移至另外一个1.5mL离心管中,并加入等体积的氯仿。
④低速涡旋5~10s并瞬时离心,转移上层水相至一新的离心管中,并向水相加入2倍体积预冷的95%乙醇,1/10体积的乙酸钠(pH4.5)和20μg的糖苷。
⑤低速涡旋5~10s并在4℃条件下以14000r/min离心15min。
⑥弃上清并用100μL预冷的80%乙醇洗涤,然后在4℃条件下以14000r/min离心10min。
⑦弃上清并在空气中干燥后用20μL TE缓冲液(pH7.5)溶解。
1.1.4接头Adaptor连接到经上述4种限制性内切酶分别消化的基因组片段上。
①连接体系:
②连接条件:16℃过夜连接。
③终止连接条件:70℃ 5min。
④每管加入75μL TE缓冲液(pH7.5),即获得4种限制性内切酶的大黄鱼基因组单酶切文库,用于启动子的克隆。
1.2采用两轮降落PCR方法扩增出大黄鱼IFNd基因启动子序列,具体步骤按GenomeWalker Universal Kit试剂盒说明书进行。具体步骤如下:
1.2.1第一轮降落PCR反应体系:
PCR程序:
1.2.2第二轮降落PCR:分别以第一轮PCR产物为模板,利用巢式接头引物AP2和基因特异引物GSP2进行,其他成分和反应体系与第一轮相同。
PCR程序:
1.3扩增得到的PCR产物连接到TaKaRa公司pMD18T-Simple vector载体进行序列测定与组装,从而获得该大黄鱼IFNd基因启动子序列。
2.大黄鱼IFNd基因启动子的活性分析
2.1含大黄鱼IFNd基因启动子片段的重组荧光素酶报告基因载体pGL3-IFNdP的构建
将大黄鱼IFNd基因启动子片段插入Promega公司荧光素酶报告基因载体pGL3-Basic中,使萤火虫荧光素酶(Luciferase)报告基因的表达受IFNd启动子控制,构建得到的重组载体命名为pGL3-IFNdP。
具体步骤:
①合成带有Xho I酶切位点的正向引物:5'-CCGCTCGAGTTCATGCTCAGGTAA-3',带有Hind III酶切位点的反向引物:5'-CCCAAGCTTAACTGCAATGCTGAAT-3'。
②用Takara公司PrimeSTAR HS DNA Polymerase聚合酶体系进行PCR扩增。
PCR反应体系为:
PCR程序如下:
③用Omega公司胶回收试剂盒进行PCR产物回收。
④回收后的PCR产物和载体pGL3-Basic分别进行Xho I/Hind III双酶切。酶切体系:
酶切条件:37℃反应过夜。
⑤用胶回收试剂盒分别回收上述经Xho I/Hind III双酶切的PCR产物和载体pGL3-Basic,并用Takara公司T4连接酶连接经双酶切的PCR产物和载体pGL3-Basic,连接体系:
反应条件:16℃反应过夜。
⑥上述连接产物转化大肠杆菌E.coli DH5α感受态细胞,经菌落PCR筛选阳性克隆,用小量质粒试剂盒提取质粒,并经测序确认启动子片段插入的正确性,从而获得含大黄鱼IFNd基因启动子片段的重组载体pGL3-IFNdP。
2.2采用双荧光素酶报告基因检测系统分析大黄鱼IFNd基因启动子的基础活性
①把状态较好的EPC细胞接种至96孔板中(1×105/孔),加入L15培养基,转入恒温培养箱28℃中过夜培养,使其贴壁并恢复至对数生长期的状态。
L15培养基配方:L15基础培养基,10%Gibco澳洲胎牛血清。
②转染前2h更换细胞培养基。转染时,1.5mL Ep管中加入30μL L15培养基、0.6μg重组质粒pGL3-IFNdP、6ng海肾荧光素酶内参报告基因载体pRL-TK以及12μL HD转染试剂(Promega)并吹打混匀后室温孵育15min共转染EPC细胞(三个平行的量)。同时,用0.6μg空载体pGL3-Basic和6ng pRL-TK共转染的EPC细胞作为对照。
48h后收集转染细胞,用双荧光素酶报告基因检测系统分别读取萤火虫荧光素酶和海肾荧光素酶的酶活性值,通过计算二者酶活性值的比值得出转染细胞中荧光素酶的相对活性。同时,以空载体pGL3-Basic和pRL-TK共同转染的EPC细胞作为对照,计算出该启动子的相对活性。荧光素酶酶活测定的方法参考Promega公司双萤光素酶报告基因检测系统说明书进行,具体步骤为:
③转染EPC细胞48h后,吸去上层培养基,用PBS缓冲液洗涤细胞2次;
④每孔加入100μL PLB裂解液(Passive Lysis Buffer,试剂盒提供)于室温下裂解细胞15min,期间轻缓晃动培养板,使其裂解完全,离心收集细胞裂解液上清;
⑤在检测管中将100μL荧光素酶测试试剂II LARII和20μL上述细胞裂解液上清混合,萤火虫荧光素酶的活性立即用化学发光检测仪Luminometer TD 20/20测量;
⑥向检测管中加入100μL Stop&Glo试剂,将上述反应猝灭,同时启动海肾荧光素酶反应,测量海肾荧光素酶活性;
⑦分别读取萤火虫荧光素酶和海肾荧光素酶的酶活性值,计算二者酶活性值的比值得出转染细胞中荧光素酶的相对活性。同时,以空载体pGL3-Basic和pRL-TK共同转染的EPC细胞作为对照,计算出该启动子的相对活性(图1)。
2.3免疫刺激实验
免疫刺激实验共实施两种方案:1)将上述重组载体pGL3-IFNdP、pRL-TK与不同剂量的poly(I:C)或细菌基因组DNA共转染EPC细胞,48h后收集转染细胞。2)上述重组载体pGL3-IFNdP、pRL-TK转染EPC细胞6h后,在细胞培养基中分别加入LPS及PGN至不同终浓度,36h后收集转染细胞。
在不同浓度LPS刺激条件下大黄鱼IFNd基因启动子的活性变化参见图3。在LPS浓度为0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.6倍、5.8倍、7.2倍、7.4倍及7.4倍。该大黄鱼IFNd基因启动子可被LPS诱导激活。*p<0.05,**p<0.01.
在不同浓度PGN刺激条件下大黄鱼IFNd基因启动子的活性变化参见图4。在PGN浓度为0ng/mL、100ng/mL、500ng/mL、1000ng/mL、2000ng/mL时,重组载体pGL3-IFNdP转染EPC细胞中的荧光素酶相对活性为空载体pGL3-Basic转染EPC细胞的4.6倍、6.1倍、7.5倍、6.6倍及5.6倍。该大黄鱼IFNd基因启动子可被PGN诱导激活。*p<0.05,**p<0.01.
用上述双荧光素酶报告基因检测系统分别计算在不同剂量或浓度的药物刺激条件下转染细胞中荧光素酶相对活性,并与未经处理转染细胞中荧光素酶相对活性相比较。
从图2和图5可以发现,不同剂量的poly(I:C)及细菌基因组DNA的刺激能显著地增强转染细胞的荧光素酶相对活性,说明了大黄鱼IFNd基因启动子可被poly(I:C)、LPS、PGN及细菌基因组DNA诱导激活。
序列表
<110> 国家海洋局第三海洋研究所
<120> 大黄鱼干扰素d基因启动子序列及其应用
<130> 1
<160> 1
<170> PatentIn version 3.5
<210> 1
<211> 1005
<212> DNA
<213> 大黄鱼< Larimichthys crocea>
<400> 1
caaagttccc acgaagtcct atcttatcta aagacacgtg tctgtcaaca ctgcagcact 1
tcagtgactc ccagccctca tatgacacag tgctgatttg tttcaggtgt tctgatgaag 61
agctggagga gaatcccctc ctgcacagac agactgcaga cactgcagca gcagcagcag 121
cttcatgctc tcaggtaaga caacgtgaaa tgtcacgcag ctctcatcaa tgaacgtcag 181
gccacagtga cggctcactt tcattttctc tgcagggaaa cttccgcttt atggctcctc 241
ttcctcattc atcaccaatc aacacaagtc atgatgcaag tcaattaaac acaaacagac 301
gtgatgacga cattaaaaca tacgtaatca tcataaatat gaaatactgt aaactgatta 361
aattcaacag ataaactaaa gtttaaatca ttatttatga aggtgatttt aattcaagga 421
tcatttacag tcattttcta tttgatctga aatattccga taataatcca taaattattg 481
aaataagtta ctcaaataac agtgagctgt gtttatcttt atgtttttaa ttttctatgt 541
tttttaaatt atgttttaac tttgctgtta ttgttgttca tgcttttatt ttgtgacatt 601
tcatattccc aggaacatta attcaaatcc gaaatgtgtg acctgggttt gagccgcgtt 661
agccacagtc cacgtcgcga ttcccacagg tgaagtttct gtgtttgttt caaatgatgt 721
tttaactttg ttgttcatgt ttttattttg tgacatttca tattcccagg aataattcaa 781
atccgaagtg tgtgggctgg tagattccca caggtggatg tgaggaaaat gaaataggtg 841
cgtcctgcta cagtataaat gagccgctgc aggtgagttt gaacacaaca cctggacaca 901
cccgactttt gccactcaaa gacactttgt ctgtttgtaa agatg 961
- 还没有人留言评论。精彩留言会获得点赞!