一种多巴胺及其衍生物聚合并交联固化的表面改性方法与流程
本发明涉及一种多巴胺及其衍生物聚合并交联固化的表面改性方法,属于复合材料制备技术领域。该方法具体涉及筛选合适的固化剂对多巴胺及其衍生物形成的聚合物进行交联固化,从而提高复合材料的表面亲水改性层在有机溶剂和酸碱性环境中的稳定性,保证改性效果长期稳定有效。
背景技术:
在保持基质材料本征性能的基础上,表面改性能够赋予其功能化的表面新性质,从而将多种材料的优点结合于一体,满足生产和生活中的需要。常用的表面改性方法有化学接枝法(自由基转移、界面交联、元素取代等)、物理复合法(颗粒掺混、表面涂覆、共混等)以及物理化学接枝法(辐射接枝、等离子处理、光诱导接枝等)。其中,化学接枝法通过改性剂与基质材料主体的化学反应而实现复合,以改变基质材料的原有分子结构为前提,而一定程度上降低了基质材料的本征性能,主要体现在机械强度变低、化学稳定性变差等方面;物理复合法,以基质材料和表面改性层之间的非共价键结合力为前提,当改性材料与基质材料性质差异较大时,二者之间的结合力较弱,改性产品的功能化表层在使用过程中容易被破坏,性能衰退明显,使用寿命短。
聚多巴胺改性是近年来受贻贝类粘附启发而发展起来的新型高效表面修饰技术,通过小分子多巴胺及其衍生物在惰性材料表面的氧化自聚,为制备高性能复合材料提供了新的方向。自2007年第一次报道以来,聚多巴胺表面改性已经在多个领域广泛研究,包括生物成像、药物探针、光催化、水处理等。聚多巴胺改性具有两大优点:1)依靠自身的氧化聚合,不会破坏基质材料的分子结构,能更好地结合多种材料的优点;2)两亲性小分子多巴胺及其衍生物为改性单体,能浸润大多数惰性材料,具有良好的适应性。除此之外,聚多巴胺改性涂层含有大量活性基团(氨基、亚氨基、酚羟基),可以作为一个中间修饰层进行二次表面改性,从而将兼容困难的不同材料结合起来。
随着应用研究的不断深入,聚多巴胺表面改性存在的局限性也逐渐被大家认识。其中,聚多巴胺在有机溶剂(或含有机溶剂的水溶液)和酸碱性环境中的稳定性较差,已经成为制约其实际应用的重要瓶颈。在聚多巴胺涂层的形成过程中,由于溶解度随着聚合过程发生显著改变,大分子高聚多巴胺将夹带大量小分子低聚多巴胺沉积在基质材料的表面。当表面改性涂层长期暴露在有机溶剂或者酸碱性环境中时,小分子低聚多巴胺由于溶解度较高,逐渐从涂层溶解进入周围的环境中,大大降低了表面改性涂层的致密程度和连续性,从而影响聚多巴胺涂层的长期稳定性,极大地限制了聚多巴胺涂层的使用范围。
综上所述,为了进一步拓宽聚多巴胺表面涂层改性技术的适用范围,更好地利用多巴胺及其衍生物,提高聚多巴胺表面改性涂层在有机溶剂(或含有机溶剂的水溶液)和酸碱性环境中的长期稳定性是必须解决的问题。为此,本发明从聚多巴胺的聚合原理及聚多巴胺涂层的形成过程出发,以聚多巴胺在有机溶剂和酸碱性环境中的失效机制为基础,筛选引入能够与聚多巴胺中的活性功能团发生反应的交联固化剂,对可溶解的小分子低聚多巴胺进行三维网状的交联固化,从本质上降低聚多巴胺在有机溶剂和酸碱环境中的溶解度,大幅提高聚多巴胺表面改性涂层的稳定性,从而保证表面改性效果长期稳定有效。
技术实现要素:
本发明的目的在于提供一种多巴胺及其衍生物聚合并交联固化的表面改性方法。该方法从聚多巴胺在有机溶剂和酸碱性环境中的失效机制出发,筛选引入交联固化剂,与聚多巴胺中的活性功能团发生反应,将可溶性小分子低聚多巴胺交联固化成不溶的三维立体网络结构,强化聚多巴胺分子之间以及聚多巴胺分子与基质材料之间的结合强度,最终制备出在机溶剂和酸碱性环境等恶劣条件下长期稳定有效的聚多巴胺表面改性涂层,极大地拓宽聚多巴胺表面涂层改性方法的使用范围。
本发明的技术方案:
一种多巴胺及其衍生物聚合并交联固化的表面改性方法,步骤如下:
1)聚多巴胺改性涂层的制备:将待改性的基质材料浸泡在含有多巴胺或其衍生物的表面改性溶液中,或将含有多巴胺或其衍生物的表面改性溶液涂敷在待改性的基质材料上,在30~70℃条件下多巴胺或其衍生物进行氧化自聚,反应时间根据表面涂层改性所需的程度进行确定;
2)聚多巴胺改性涂层的交联固化:对步骤(1)制备的经聚多巴胺表面改性的基质材料进行洗涤,除去表面残余未聚合的多巴胺或其衍生物,再将改性后的基质材料浸泡在交联固化溶液中,或将交联固化溶液涂覆在改性后的基质材料上,在30~90℃条件下反应3~6h,完成多巴胺或其衍生物的聚合涂层的交联固化,提高改性涂层在有机溶剂和酸碱性环境中的长期稳定性。
所述的交联固化剂,包括多聚甲醛、甲醛、乙醛、戊二醛、多胺、环氧烯烃,或者上述多种固化剂的混合物。
所述多巴胺的衍生物,可以是左旋多巴胺、二羟基苯基丙基甲基丙烯酰胺、氢醌、邻苯二酚,或者上述衍生物的混合物。
所述含有多巴胺或其衍生物的表面改性溶液,可以只含有多巴胺及其衍生物的一种,也可以含有多种多巴胺及其衍生物,改性溶液可以是碱性体系,也可以是中性体系或者酸性体系。
所述的基质材料,可以是无机材料(二氧化硅、石墨、炭、陶瓷、金属氧化物等物质)、有机材料(高分子聚合物、生物大分子等物质)和金属材料,其形态可以是颗粒、丝、棒、板、网和多孔膜等等。
本发明的有益效果:通过交联固化剂与聚多巴胺中的活性功能团反应,将可溶性小分子低聚多巴胺交联固化成不溶的三维立体网络结构,强化聚多巴胺分子之间以及聚多巴胺分子与基质材料之间的结合强度,制备出在机溶剂和酸碱性环境等恶劣条件下长期稳定有效的聚多巴胺表面改性涂层,极大地拓宽聚多巴胺表面涂层改性方法的使用范围。
附图说明
图1为PVDF原膜、聚多巴胺改性PVDF膜、聚多巴胺/多聚甲醛固化改性PVDF膜的红外光谱图。
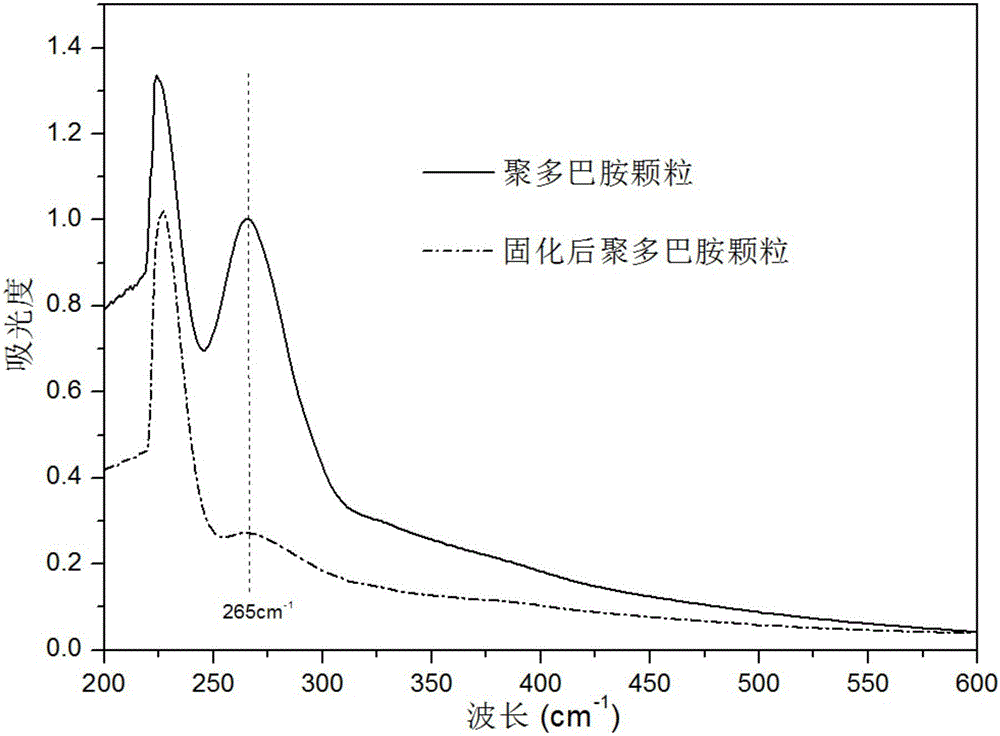
图2为聚多巴胺颗粒以及聚多巴胺/多聚甲醛固化颗粒在DMSO溶液中溶解情况的紫外光谱对比分析图。
图3为聚多巴胺改性PVDF膜、聚多巴胺/多聚甲醛固化改性PVDF膜在有机溶剂DMSO、NMP、DMF中以及1M的NaOH碱性溶液中聚多巴胺保留量随时间的变化图。
图4为聚左旋多巴胺改性PVDF膜、聚左旋多巴胺/甲醛固化改性PVDF膜在HCl溶液(24h)中聚多巴胺保留量随HCl浓度的变化图。
图5(a)为聚左旋多巴胺改性PVDF膜在2M的HCl溶液中浸泡24h后的扫描电镜图。
图5(b)为聚左旋多巴胺/甲醛固化改性PVDF膜在2M的HCl溶液中浸泡24h后的扫描电镜图。
具体实施方式
以下结合附图和技术方案,进一步说明本发明的具体实施方式。
实施例1
采用多巴胺(浓度为2g/L)为表面改性物质,在弱碱性缓冲液中对聚偏二氟乙烯(PVDF)微孔膜进行表面涂覆,然后将涂覆后的复合膜用多聚甲醛(浓度为10g/L)进行固化,得到亲水改性后的复合膜。
操作步骤如下:
1)在Tris缓冲溶液(50ml,pH=8.5)中加入0.1g多巴胺,制备浓度为2g/L的多巴胺溶液;加入0.02g碘酸钠,充分溶解成多巴胺的表面改性溶液;
2)将正辛醇浸泡3h的PVDF微孔膜(直径90mm,孔径0.22μm)浸入第(1)步配制的表面改性溶液中,反应温度为40℃,反应时间为8小时;
3)将完成聚多巴胺涂覆的PVDF微孔膜取出,去离子水洗涤除去未聚合的多巴胺,然后在烘箱中加热至60℃干燥2h,得到聚多巴胺涂覆的复合膜。
4)取0.5g多聚甲醛充分溶解于盐酸溶液(50ml,pH=2)中,配制成多聚甲醛交联固化溶液。
5)将第(3)步制备的聚多巴胺涂覆的PVDF微孔膜浸泡在第(4)步配制的多聚甲醛交联固化溶液中,反应温度为90℃,表面固化时间为3h,得到聚多巴胺/多聚甲醛固化改性的PVDF微孔膜。
6)将第(3)步制备的聚多巴胺改性PVDF微孔膜和第(5)步制备的聚多巴胺/多聚甲醛固化改性PVDF微孔膜分别置于20ml的DMSO、DMF、NMP以及1M的NaOH溶液中测试聚多巴胺涂层的稳定性。
图1展示了PVDF微孔膜经聚多巴胺改性、聚多巴胺/多聚甲醛固化改性后官能团的变化。由图中红外光谱中特征吸收峰可知,聚多巴胺成功涂覆到PVDF微孔膜上。聚多巴胺经过多聚甲醛交联固化后,功能基团未发生明显变化,表明固化过程所消耗的聚多巴胺中的功能基团很少,对聚多巴胺涂层的改性效果不会造成显著影响。
图2中紫外光谱反映了聚多巴胺颗粒以及聚多巴胺/多聚甲醛固化颗粒在DMSO溶液中的溶解情况。多聚甲醛的交联固化成功地将小分子低聚多巴胺构筑成具有立体网络结构的大分子聚合物,显著降低了聚多巴胺在极性有机溶剂DMSO中的溶解度。
步骤(6)实验的结果如图3所示,进一步说明了多聚甲醛交联固化显著提高了聚多巴胺的稳定性。未固化的聚多巴胺改性涂层,在有机溶剂中浸泡24h后溶解损失了10~20%,在碱性溶液中浸泡24h后溶解损失了80%以上;经过多聚甲醛交联固化的聚多巴胺改性涂层,在极性有机溶剂中浸泡24h后溶解损失小于5%,在碱性溶液中浸泡24h后溶解损失小于20%。上述结果表明,多聚甲醛交联固化后的聚多巴胺改性涂层,在有机溶剂和酸碱性环境中的稳定性得到显著提高。
实施例2
采用左旋多巴胺(浓度为3g/L)为表面改性物质,在弱碱性缓冲液中对聚偏二氟乙烯(PVDF)微孔膜进行表面涂覆,然后将涂覆后的PVDF微孔膜用甲醛溶液(体积含量10%)进行交联固化,得到亲水改性后的复合膜。
操作步骤如下:
1)在Tris缓冲溶液(50ml,pH=8.5)中加入0.15g左旋多巴胺,制备浓度为3g/L的左旋多巴胺溶液;加入0.02g碘酸钠,充分溶解成左旋多巴胺的表面改性溶液;
2)将正辛醇浸泡3h的PVDF微孔膜(直径90mm,孔径0.22μm)浸入第(1)步配制的表面改性溶液中,反应温度为40℃,反应时间为12小时;
3)将完成聚左旋多巴胺涂覆的PVDF微孔膜取出,去离子水洗涤除去未聚合的左旋多巴胺,然后在烘箱中加热至60℃干燥2h,得到聚左旋多巴胺涂覆的复合膜。
4)取5ml甲醛充分溶解于盐酸溶液(50ml,pH=2)中,配制成甲醛交联固化溶液。
5)将第(3)步制备的聚左旋多巴胺涂覆的PVDF微孔膜浸泡在第(4)步配制的甲醛交联固化溶液中,反应温度为70℃,表面固化时间为6h,得到聚左旋多巴胺/甲醛固化改性的PVDF微孔膜。
6)将第(3)步制备的聚左旋多巴胺改性PVDF微孔膜和第(5)步制备的聚左旋多巴胺/甲醛固化改性PVDF微孔膜,分别置于20ml的2M、4M、6M的HCl溶液中,测试聚左旋多巴胺涂层在酸性环境中的溶解损失量。
图4为聚左旋多巴胺改性PVDF膜、聚左旋多巴胺/甲醛固化改性PVDF膜在HCl溶液(24h)中聚多巴胺保留量随HCl浓度的变化图。结果表明,甲醛交联固化后的聚左旋多巴胺涂层,在酸性环境中的稳定性显著提高,在6M的HCl溶液中浸泡24h后,溶解损失量不到5%。即使在6M的HCl溶液中浸泡2个月,其长期溶解损失量也只有5%左右。
进一步从微观尺度上观察甲醛交联固化对聚左旋多巴胺改性涂层在酸性环境中稳定性的影响,如图5所示。图5(a)为聚左旋多巴胺改性PVDF膜在2M的HCl溶液中浸泡24h后的扫描电镜图。图5(b)为聚左旋多巴胺/甲醛固化改性PVDF膜在2M的HCl溶液中浸泡24h后的扫描电镜图。未交联固化的膜,聚左旋多巴胺出现明显的溶解损失和脱落,膜表面的聚左旋多巴胺涂层凸起尺度不均匀、排列不连续;甲醛交联固化后的膜,聚左旋多巴胺涂层没有明显的溶解损失和脱落痕迹,膜表面的聚左旋多巴胺涂层排列致密、尺均匀。显然,甲醛交联固化,极大地提高了聚左旋多巴胺改性涂层在酸性环境中的长期稳定性。
- 还没有人留言评论。精彩留言会获得点赞!