猪肌抑素基因编辑位点864‑883及其应用的制作方法
本发明属于生物技术和生物工程领域,具体涉及猪肌抑素基因编辑位点及其应用。
背景技术:
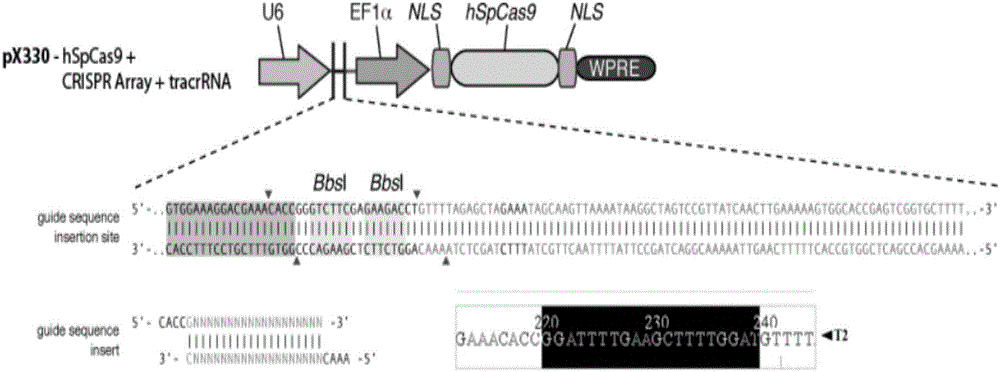
:基因转移技术根据外源基因整合的位点特异性可分为两大类,一类是非定点的,即导入的外源基因在靶细胞基因组中整合的位点一般是随机的,包括显微注射、电穿孔、磷酸钙沉淀、逆转录病毒载体感染等:另一类则是定点的,即外源基因导入靶细胞后整合到预先确定的位点或对细胞内靶位点进行定点修饰,这就是依赖于同源重组的基因打靶技术。将外源基因精准敲入猪体细胞的基因组中,一直都是极为困难的,因为早期仅基于同源重组的基因打靶技术效率极低,应用受到很大的限制。直到近年来人工核酸内切酶(engineeredendonuclease,EEN)的出现,才彻底改变了这一现状。锌指核酸内切酶(zincfingerendonuclease,ZFN)是第一代人工核酸内切酶。锌指是一类能够结合DNA的蛋白质,很多转录因子中都含有锌指结构。ZFN是将锌指蛋白的DNA结构域和FokI核酸内切酶中的非特异性的DNA切割结构域组合形成的一种嵌合体蛋白,其既具有锌指蛋白结合DNA的能力又具有核酸内切酶FokI切割DNA双链的能力(KimYG,ChaJ,ChandrasegaranS.Hybridrestrictionenzymes:zincfingerfusionstoFokIcleavagedomain.ProcNatlAcadSciUSA,1996,93(3):1156-1160.)。利用ZFN可以在各种复杂基因组的特定位置制造DNA的双链切口。到目前为止,ZFN已经成功应用于猪、牛、大鼠、小鼠、斑马鱼、家蚕、果蝇、海胆、拟南芥、烟草、玉米等生物(UrnovFD,RebarEJ,HolmesMC,etal.Genomeeditingwithengineeredzincfingernucleases.NatRevGenet,2010,11(9):636-646.)。但是,由于ZFN制备流程复杂,成本昂贵,而且其技术专利被少数几家商业公司所控制,所以推广应用十分有限。2009年,科学家将一种水稻的致病菌(Xanthamonas)编码的类转录激活因子效应物(transcriptionactivator-likeeffector,TALE)与DNA的碱基对应关系解密(MoscouMJ,BogdanoveAJ.AsimpleciphergovernsDNArecognitionbyTALeffectors.Science,2009,326(5959):1501;BochJ,ScholzeH,SchornackS,etal.BreakingthecodeofDNAbindingspecificityofTAL-typeIIIeffectors.Science,2009,326(5959):1509-1512.)。第二代人工核酸酶——类转录激活因子效应物核酸酶(transcriptionactivator-likeeffectornuclease,TALEN)应运而生。和ZFN相似,TALEN主要由TALE蛋白的DNA结合结构域和FokI核酸内切酶结构域组合而成,TALE蛋白含有多个33-35个氨基酸组成的重复肽段,而每一个肽段都能够识别一个碱基。2010年,首次报道TALEN蛋白在酵母中应用成功(LiT,HuangS,ZhaoX,etal.ModularlyassembleddesignerTALeffectornucleasesfortargetedgeneknockoutandgenereplacementineukaryotes.NucleicacidsRes,2011,39(14):6315-6325.)。之后,TALEN在植物、小鼠、斑马鱼、猪、牛等动植物中得到了迅速的推广与应用(JoungJK,SanderJD.TALENs:awidelyapplicabletechnologyfortargetedgenomeediting.NatRevMolCellBiol,2013,14(1):49-55.)。TALEN不仅可以像ZFN一样对复杂的基因组进行精细的修饰,而且其构建更加简单,特异性也更高,因此TALEN出现不久就在很大程度上替代了ZFN。2012年,TALEN被《科学》(Science)杂志评为十大科学突破之一(Breakthroughoftheyear.Therunners-up.Science,2012,338(6114):1525-1532.)。无论是ZFN还是TALEN,这两种人工核酸酶都是嵌合体,都是由经过人工设计的、序列特异性的DNA结合元件和非特异性的DNA切割结构域结合而成的。TALEN与ZFN的作用机制都是先对DNA双链分子靶序列进行切割,形成DNA双链断裂(double-strandbreak,DSB),然后DSB激活细胞自身的DNA损伤修复机制,从而对该DNA进行定点改造。2013年初,一种继ZFN和TALEN之后的第三代基因定点编辑技术clusteredregularlyinterspacedshortpalindromicrepeats(CRISPR)/CRISPR-associated(Cas)闪亮登场。CRISPR/Cas系统主要是基于细菌的一种获得性免疫系统改造而成,细菌利用此系统可以特异性识别入侵的外源DNA并利用Cas蛋白对目标DNA进行断裂和降解,从而抵抗病毒感染。CRISPR/Cas9基因编辑系统的工作原理:crRNA(CRISPR-derivedRNA)通过碱基互补配对与tracrRNA(trans-activatingRNA)结合形成tracrRNA/crRNA复合物,此复合物引导核酸酶Cas9蛋白在与crRNA配对的序列靶位点剪切双链DNA,产生DNA双链断裂(DSB),然后DSB激活细胞内的DNA损伤修复机制,通过两种方式对DNA进行修复,即非同源末端连接(non-homologousendjoining,NHEJ)或者同源重组修复(homology-directedrepair,HDR),从而达到定点改造基因组(包括内源基因的敲除或外源基因的定点敲入)的目的。NHEJ是一种非保真的、容易出现遗传突变的修复机制,往往使得核酸链被剪切的区域发生基因突变,导致编码的基因Knockdown或Knockout。而HDR则是一种高保真的修复机制,不容易出现突变,往往通过供体DNA与基因组DNA之间的同源重组造成靶位点的纠正或者靶向插入外源基因Knockin。CRISPR/Cas9技术拥有ZFN和TALEN所不具备的独特优势:1.CRISPR/Cas9系统只需合成一个sgRNA(编码sgRNA的序列不超过100bp)即可实现对基因的特异性修饰,所以操作起来更加简单快捷。2.CRISPR/Cas9系统中,sgRNA靶向序列和基因组序列必须完全匹配,Cas9才能对DNA进行剪切,因此其靶向精确性性更高。3.CRISPR/Cas9系统基因修饰率更高,对基因调控方式更多样(敲除、插入、抑制、激活等)。4.CRISPR/Cas9系统的实验周期更短,可节省大量时间和成本。6.CRISPR/Cas9系统更容易得到纯合子突变体,而且可以在不同的位点同时引入多个突变(MussolinoC,CathomenT.RNAguidesgenomeengineering.NatBiotechnol,2013,31(3):208-209;Wang,H.,etal.(2013)."One-stepgenerationofmicecarryingmutationsinmultiplegenesbyCRISPR/Cas-mediatedgenomeengineering."Cell153(4):910-918.)。CRISPR/Cas9技术自问世以来,就有着其它基因编辑技术无可比拟的优势,在短短两三年之内,技术不断改进,发展成为生物学领域最受青睐的研究工具之一。基因打靶是通过同源重组技术将外源基因定点整合进靶细胞基因组上某一确定的位点,以达到定点修饰改造染色体上某一基因的目的,具有技术成熟、修饰准确、效果稳定等优点。基因打靶技术目前已被广泛认为是一种理想的特定修饰与改造生物体遗传物质的最佳方法,其中包括了多种不同的基因敲除和敲入系统,特别是条件性和诱导性基因打靶系统的建立,使得对基因在时间和空间上的靶位修饰更加明确、效果更加精确可靠。将CRISPR/Cas9技术和基因打靶联合起来,核酸内切酶Cas9定点切割靶DNA后产生DNA断口,再利用打靶载体的同源臂进行精确的同源重组,相对提高了该位点的短片段同源重组的效率。本人于2016年5月12日检索PubMed,以关键词“CRISPR/Cas9”查询,检出文献1680篇,其中2011年仅发表1篇,2012年发表3篇,2013年发表78篇,2014年发表316篇,2015年发表718篇,2016年截止到2016年5月12日已发表564篇。以关键词“myostatinCRISPR/Cas9”查询,检出文献为5篇,其中仅有一篇文章是利用CRISPR/Cas9系统对猪MSTN基因进行高效突变(Wang,K.,etal.(2015)."EfficientGenerationofMyostatinMutationsinPigsUsingtheCRISPR/Cas9System."SciRep5:16623.)。本研究是对猪MSTN第3外显子进行打靶,根据打靶后统计结果表明,该位点打靶效率高达86.7%,与基于胚胎干细胞(ES)的同源重组技术即俗称的“传统的基因打靶技术”相比,打靶效率有了极大的提高。肌肉生成抑制素(myostatin,MSTN),又称GDF-8,属于转化生长因子超家族。MSTN基因在哺乳动物进化过程中相当保守,基因编码区均含有2个内含子和3个外显子,仅有若干个单核苷酸的突变或错义突变。MSTN基因编码一种能够对肌肉生长起负调控作用、化学本质为糖蛋白的生长因子,可影响畜禽产肉率、改善肉质性状,并广泛分布在骨骼肌中。因此,在畜牧业上,MSTN基因的多态可作为选择肌肉性状和胴体性状的分子标记。随着CRISPR/Cas9技术的不断改进,使得对猪MSTN基因进行编辑变得更为简单快捷,该技术的应用范围包括:一方面,对猪MSTN基因进行完全性基因敲除,可培育出肌肉发达的瘦肉型猪,从而提高了养猪业的经济效益;另一方面,进行基因定点插入,点突变研究和人源化研究。对进一步研究MSTN基因的功能及其与某些疾病的相互联系具有十分重大的意义。因此,本发明的目的在于鉴定MSTN基因的高效编辑位点,并分析其打靶效率,为构建MSTN基因内源性敲除或定点敲入外源基因的转基因猪提供新途径。技术实现要素:本申请以pX330-U6-Chimeric_BB-CBh-hSpCas9载体为骨架构建插入有猪肌抑素基因(MSTN)编辑靶位点(864-883)的Cas9gRNA表达载体T21,并将其与带有靶位点附近同源臂的打靶载体dT2按照一定的摩尔比例转染猪肾PK15细胞,以G418筛选单克隆转基因细胞系。为方便筛选阳性克隆,打靶载体dT2设计有绿色荧光蛋白(GFP)和红色荧光蛋白(RFP)双荧光蛋白,如果发生精确的同源重组,而非随机整合的话,则细胞应该只发绿色荧光。同时,打靶载体dT2含有neo基因,可用G418辅助筛选。本发明的目的是:提供一个可高效敲除猪肌抑素基因(MSTN)的编辑位点(864-883)。该编辑位点可用于Cas9介导的猪MSTN基因打靶,将肌抑素功能失活,创建肌抑素缺失转化体,操作步骤包括:(1)构建含有编辑靶位点(864-883)的Cas9gRNA表达载体T21和带有双荧光蛋白的打靶载体dT2;(2)将上述2种载体按照一定摩尔比例转染猪肾PK15细胞;(3)G418加压筛选仅发绿色荧光的单克隆转基因细胞系;(4)PCR鉴定同源重组的真实性;(5)测序验证重组边界的准确序列。利用本发明提供的编辑位点(864-883),可以在核酸内切酶Cas9的介导下以较高效率进行双链断裂,从而提高打靶载体和目的基因的同源重组效率,将带有选择性标记的外源基因定点整合于猪基因组内。该编辑位点(864-883)位于猪肌抑素基因(MSTN)的3号外显子区,可高效的敲除MSTN基因并定点整合有neo和EGFP选择性标记。本发明将提供一种方便、高效的策略来构建MSTN基因敲除的转基因细胞系,对促进内源基因敲除或外源基因定点整合技术在转基因猪研究和生产中的应用具有十分重要的作用。除非特别指出或是单独定义,本文所使用的科学和技术术语具有本发明所属领域技术人员共知的、无歧义的相同含义。本文所提及的所有已公开的专利申请和参考文献均已通过完整引用的方式并入本申请中。附图说明图1编辑位点(864-883)在猪基因组上的准确位置猪肌抑素基因MSTN编辑位点T2是一段长23bp的已知DNA序列,序列为:GGATTTTGAAGCTTTTGGATGGG,该位点在猪基因组的准确定位是15号染色体MSTN编码区3号外显子上。图2插入有引导序列的Cas9gRNA表达载体示意图及编辑位点序列。上图为Cas9gRNA表达载体示意图,以pX330-U6-Chimeric_BB-CBh-hSpCas9载体为骨架,带有hSpCas9基因、CRISPRArray和tracrRNA序列的一个表达载体。下图所示为编辑位点(864-883)序列。图3dT2质粒及酶切的电泳图。M为1kbDNAladder(TaKaRaM1181/M1182),右侧依次为未经酶切的质粒、经NsiⅠ和XhoⅠ双酶切后的产物。可见,原始质粒及酶切后产物与预期一致。图4基因打靶示意图。如果为同源重组,则neo和EGFP选择性标记整合至MSTN的864-883位点。检测打靶后的整合情况分别用T2-Up和MKR这对引物检测5’arm(预期PCR产物的长度约为759bp),用EGFP-QF1和T2-Down这对引物检测3’arm(预期PCR产物的长度约为1.7kb)。图5电穿孔转染后可能阳性的克隆斑的显微照片。由左往右,依次为转染后可能阳性的PK-15克隆斑在明场(bright)、绿色荧光蛋白(GFP)激发波长和红色荧光蛋白(RFP)激发波长下的显微照片。明场下可见PK-15细胞的大小、形态均正常。绿色荧光蛋白(GFP)激发波长下可见该克隆斑发出明亮的绿色荧光,而红色荧光蛋白(RFP)激发波长下无明显可见的红色荧光,证明neo和EGFP选择性标记已经整合至基因组中,而RFP没有整合进去,很可能就是所需要的阳性克隆。图65’同源重组边界的确认。在整合位点的5′arm设计跨界引物,扩增猪基因组DNA与外源基因neo之间的片段,反证外源基因的插入位置。1号泳道为DL2000DNALadder,2号泳道和3号泳道为未修饰猪基因组DNA为模板,4号泳道为水模板,5-9号泳道为50ng克隆斑所提的基因组DNA为模板。电泳及测序结果显示在所挑选的克隆斑即转基因细胞系中,PCR扩增出759bp的特异片段,而在所有阴性对照中均无该片段。证明外源基因的插入位置与理论分析相符。图73’同源重组边界的确认在整合位点的3′arm设计跨界引物,扩增猪基因组DNA与外源基因EGFP之间的片段,反证外源基因的插入位置。1号泳道为DL2000DNALadder,2号泳道和3号泳道为未修饰猪基因组DNA为模板,4号泳道为水模板,5-9号泳道为50ng克隆斑所提的基因组DNA为模板。电泳及测序结果显示在所挑选的克隆斑即转基因细胞系中,PCR扩增出1701bp的特异片段,而在所有阴性对照中均无该片段。证明外源基因的插入位置与理论分析相符。图8打靶效率统计表经过G418筛选后的细胞,逐渐形成一个个独立的克隆斑,挑选只发绿色荧光的单克隆至12孔板上继续培养,待细胞数量足够多时,用胰酶消化后取部分提基因组供检测用。最后,统计表明:只发绿色荧光的单克隆有30个,其中有26个克隆经检测发生了同源重组即选择性标记的外源基因整合至预期的T2位点,打靶效率高达86.7%。这充分表明该编辑靶位点T2可以高效敲除MSTN基因。具体实施方式本发明是以pX330-U6-Chimeric_BB-CBh-hSpCas9载体为骨架构建插入有猪肌抑素基因(MSTN)编辑靶位点T2的Cas9gRNA表达载体T21,并将其与带有靶位点附近同源臂的打靶载体dT2按照一定的摩尔比例转染猪肾PK15细胞,以G418辅助筛选,最后仅挑选只发绿色荧光的单克隆并进行PCR检测和测序分析以确认5’arm和3’arm是否插入到预期的靶位点。以下结合实施例和附图对本发明做详细的阐释。需要指出的是,本实施例仅用于解释本发明,而非对本发明的权利要求做出限制或限定。实施例1插入有猪肌抑素基因(MSTN)编辑靶位点T2的Cas9gRNA表达载体T21的构建(1)主要试剂与材料来源pX330-U6-Chimeric_BB-CBh-hSpCas9(MammalianExpression,CRISPRhumanizedS.pyogenesCas9)载体购自Addgene,打靶载体dT2为本实验室自行设计。AgeI和EcoRI限制性内切酶均购自Fermentas。重组载体按照北京天根生物科技有限公司提供的小提中量超纯质粒抽提试剂盒说明书进行。DNA割胶回收试剂盒购自Qiagen公司。测序委托深圳华大基因有限公司完成。(1)Cas9gRNA表达载体是以购买的pX330-U6-Chimeric_BB-CBh-hSpCas9载体为骨架,然后按照固定的oligo设计模式插入所需的引导序列即可。引导序列5’CACCGGATTTTGAAGCTTTTGGATGGG3’和5’AAACCCCATCCAAAAGCTTCAAAATCC3’及测序引物Cas9-S:5’gagggcctatttcccatgattcc3’(位于U6启动子上游),均由上海英骏生物科技有限公司合成,以干粉形式分装、运输、保存。引物用灭菌后的ddH2O稀释为10μmol/L的溶液,-20℃保存备用。(1)操作步骤。-限制性内切酶AgeI和EcoRI双酶切质粒pX330-U6-Chimeric_BB-CBh-hSpCas9载体,反应体系如下:成分初始浓度用量终浓度BufferTangoTM10×3μl1×AgeI10units/μl1μl0.33unit/μlEcoRI10units/μl1μl0.33unit/μlpX330质粒1000ng/μl3μl100ng/μl总体积30μl在冰上将以上成分依次加入,充分混匀。放在37℃水浴锅中酶切过夜。-按照Qiagen公司DNA回收试剂盒的步骤回收上述酶切产物。-将回收的酶切产物连接,反应体系如下:在冰上将以上成分依次加入,充分混匀。放在37℃水浴锅中连接过夜。结束后置于冰上,用于转化。-转化大肠杆菌DH5α,转化体系如下:成分用量连接产物10μlDH5α感受态细胞100μl总体积110μl将以上成分混匀,冰浴30min;42℃热激90s;加入不含有氨苄抗生素的液体LB培养基500μl,置于37℃摇床以200rpm/min的转速复苏45min;在台式离心机上以8000rpm/min的转速离心,将菌体沉淀下来,然后去除450μlLB培养基;将剩余的100μl样品涂布在。氨苄青霉素LB平板上,置于37℃温箱过夜培养16h。挑取细菌单克隆送深圳华大测序。实施例2电穿孔转染后猪转基因细胞系的筛选(1)主要试剂与材料来源猪肾PK15细胞系购自ATCC。无内毒素质粒制备按照北京天根生物科技有限公司提供的小提中量超纯质粒抽提试剂盒说明书进行。G418抗生素购自Sigma。DMEM、DPBS、胎牛血清、DMSO购自Invitrogen。电转缓冲液实验室自行配制。BTX2001细胞电融合仪购自美国BTX公司。(2)操作步骤-细胞铺板:电穿孔前一天,将PK15细胞用胰酶消化为单细胞悬液后,按照0.25-1×106细胞/孔铺于6孔板,生长过夜。-消化细胞:根据细胞密度和数量取适量的胰蛋白酶消化收集贴壁细胞(对于PK15,一般6孔板需要加入0.25%胰酶溶液0.5-1mL/孔,39℃培养箱孵育5-10min,在显微镜下观察到有少量细胞开始脱落即加入含血清的培养基终止消化),反复吹打使贴壁细胞完全脱离下来并分散成单细胞。-收集、重悬细胞:将细胞悬液收集到洁净无菌的离心管中,先用DPBS重悬细胞,2400rpm离心3-5min,弃上清,再用电转缓冲液将细胞重悬,再次离心(为了确保将细胞彻底洗涤干净,可以用电转缓冲液洗细胞两次)。弃上清,加入适量电转缓冲液和质粒,轻轻吹打使细胞、质粒和电转液充分混匀。-电击:提前开启电转仪预运行1-2分钟并设置好电压、电击时间和电击次数等参数(100V,3ms,1pulse),吸取适量的细胞混合液加入洁净的电击杯(电击杯直径10mm对应样品总体积50μL)尽量混匀,避免气泡,然后将电击杯放入电击卡槽里,放置电击杯时一定要使电击杯金属面和卡槽电极紧贴,关上盖子,按Automaticstart进行电击。-培养:电击完成后可见wait指示灯停止闪烁后,取出电击杯,用细长的吸头吸出样品转入提前预热好的非选择性培养基内。做好相关的标记,将培养板放入39℃、5%CO2培养箱培养,关闭仪器电源并清洗电击杯(清水-蒸馏水-酒精-干燥)。培养24小时后,观察细胞生长情况,计数,统计转染效率,拍照记录。-G418筛选:培养24h后,将贴壁良好的细胞用胰酶消化为单细胞悬液,按照1:20的密度分盘,传至10cm培养皿,逐渐根据细胞死亡情况调整G418的添加量,持续培养直到单个细胞成长为肉眼可见的克隆斑。-克隆斑分离采用胰酶点消化法分离单克隆细胞。将2μl胰酶吸到单克隆细胞上,消化30s后轻轻吸打,将细胞吸起,传至12孔板中。12孔板培养基内含有100μg/ml的G418,用以维持单克隆细胞的生长。待细胞蔓延整个6孔板后,即可冻存、采样,用于后续分析。附:电转缓冲液配方:实施例3选择性标记插入位置的确认与打靶效率统计分析(1)主要试剂与材料来源TaKaRaLATaq及其配套的缓冲液(10×buffer),dNTP,MgSO4均购自东洋纺生物科技有限公司。哺乳动物基因组DNA提取试剂盒购自康为世纪。测序委托深圳华大基因有限公司完成。检测阳性克隆5’arm的一对特异性引物为T2-Up和MKR,预期PCR产物的长度约为759bp,检测3’arm的一对特异性引物为EGFP-QF1和T2-Down,预期PCR产物的长度约为1.7kb。引物由上海英骏生物科技有限公司合成,以干粉形式分装、运输、保存。引物用灭菌后的ddH2O稀释为10μmol/L的溶液,-20℃保存备用。(2)操作步骤-按照康为世纪提供的哺乳动物基因组DNA提取试剂盒说明书制备单克隆细胞系的基因组DNA,电泳检测其完整性,紫外分光光度计测定浓度和纯度,将浓度调整至20ng/μl。-5’armPCR检测:PCR的反应体系为:在冰上将以上成分依次加入,充分混匀。放入PCR仪中按照下述条件运行:-将PCR反应产物电泳并委托深圳华大基因有限公司测序,确认选择性标记基因是否在编辑靶位点的5’arm区正确整合。-3’armPCR检测:PCR的反应体系为:在冰上将以上成分依次加入,充分混匀。放入PCR仪中按照下述条件运行:-将PCR反应产物电泳并委托深圳华大基因有限公司测序,确认选择性标记基因是否在编辑靶位点的3’arm区正确整合。-统计打靶效率:经过G418筛选后的细胞,逐渐长成一个个独立的克隆斑,挑选只发绿色荧光的单克隆至12孔板上继续培养,待细胞数量足够多时,用胰酶消化后取部分提基因组供PCR检测用,剩下部分细胞若为阳性则冻存。实验中一共挑选到30个只发绿色荧光的单克隆,其中有26个克隆经检测发生了同源重组即选择性标记的外源基因整合至预期的T2位点,则统计的打靶效率为86.7%。这充分表明该编辑靶位点T2可以高效敲除MSTN基因,且外源的选择性标记基因可经同源重组定点整合到预期设计的编辑靶位点上。SEQUENCELISTING<110>湖北省农业科学院畜牧兽医研究所<120>猪肌抑素基因编辑位点864-883及其应用<130><160>4<170>PatentInversion3.3<210>1<211>23<212>DNA<213>猪<400>1ggattttgaagcttttggatggg23<210>2<211>27<212>DNA<213>人工序列<400>2caccggattttgaagcttttggatggg27<210>3<211>27<212>DNA<213>人工序列<400>3aaaccccatccaaaagcttcaaaatcc27<210>4<211>23<212>DNA<213>人工序列<400>4gagggcctatttcccatgattcc23当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1