苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物的合成方法与流程
本发明涉及一种苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物的合成方法。
背景技术:
天然卟啉化合物在自然界中以多种形式广泛存在于动植物体内,承担着与生命活动密切相关的工作,人类合成多种卟啉化合物以模拟这些生命活动。1936年,Paul Rothemund等人以吡咯和醛类化合物作为反应物,吡啶作为反应溶剂,在密封环境中控制温度在90-95℃之间反应30小时首次合成了卟吩,到了1941年,他又用等摩尔量的苯甲醛和吡咯作为反应物,在吡啶溶剂中密闭加热合成出四苯基卟啉。此方法开创了卟啉合成的先河。此后,人们在此基础上开创了多种合成卟啉化合物的方法,主要有Adler法、Lindsey法、郭灿城法、[2+2]法和微波诱导法等方法。
在研究过程中人们发现部分卟啉化合物是一种盘状液晶化合物,其母体卟吩是一个共轭平面环,相当于盘状液晶中的盘状中心,且卟啉化合物在可见光范围内存在吸收光谱,具有良好的光和热稳定性;而苯并菲衍生物是一种拥有平面结构的多环芳烃,是一类非常重要的盘状液晶材料,也具有良好的光学性质和热稳定性。在盘状液晶化合物的研究过程中,有关盘状液晶低聚物的报道远少于盘状液晶小分子和盘状液晶高聚物,但是相较于盘状液晶单分子化合物,盘状液晶低聚物具有更为宽泛的液晶中间相;同时由于盘状液晶低聚物中的盘状分子依然能够通过π-π作用力堆集成柱状相,所以它又比盘状液晶高聚物拥有更好的电荷传输能力。
本发明利用两个苯并菲单元和一个卟啉单元通过柔性桥链连接形成三聚体化合物-苯并菲癸烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物,其合成方法简便,在可见光区域具有强吸收。此类化合物未来可以应用在有机太阳能电池、有机光伏材料、液晶材料等分子器件中。
技术实现要素:
本发明的目的是提供一种苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物的合成方法。
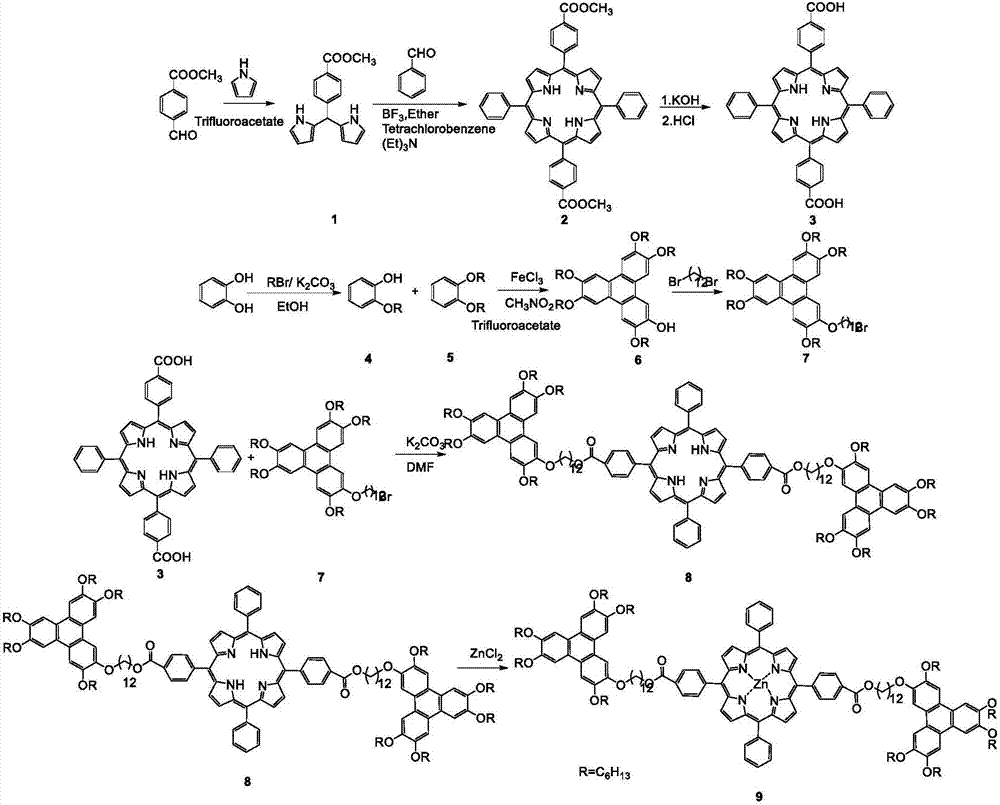
本发明合成路线分为以下三个部分:第一部分先以对甲酰基苯甲酸甲酯和吡咯为原料合成二吡咯,然后与苯甲醛反应合成卟啉酯,卟啉酯水解、酸化得到卟啉酸;第二部分在三氯化铁的氧化作用下邻己烷氧基苯酚和邻二己烷氧基苯发生偶联反应生成单羟基五己烷氧基苯并菲,再以单羟基五己烷氧基苯并菲与1,12-二溴十二烷为原料合成具有一个ω-溴支链的烷氧基苯并菲;第三部分将上述两部分得到的中间体合成烷氧基桥连苯并菲-卟啉-苯并菲三元化合物后与金属盐在N,N-二甲基甲酰胺和三氯甲烷的混合溶剂中进行反应,进而得到金属配合物。
苯并菲及其衍生物拥有良好的液晶性质,具有一维方向上的电荷传输和能量迁移性能,且合成苯并菲类化合物的原料易得,产品易于提纯;而卟啉衍生物分子性质和结构稳定,具有良好的荧光性和芳香性,卟啉金属配合物拥有更加独特的光学性质。在本发明中利用两个苯并菲单元和一个卟啉单元通过柔性桥链连接形成三聚体化合物-苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物,此类化合物未来可以应用在有机太阳能电池、有机光伏材料、液晶材料等分子器件中。
附图说明
图1为本发明苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物的结构式。
图2为本发明合成路线的化学反应方程式。
图中:R=C6H13。
具体实施方式
实施例:
实施例中所使用的化学试剂和溶剂均为分析纯。
(1)二吡咯(化合物1)的合成:
取0.22毫升三氟乙酸室温下避光滴加至35毫升含有6克对甲酰基苯甲酸甲酯的吡咯溶液中,氮气氛围下滴加30分钟完毕,接着室温下避光反应4小时后,加入400毫升二氯甲烷,反应混合物用100毫升0.1mol/L的氢氧化钠水溶液洗涤三次,再用100毫升双蒸水洗涤三次,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比4:1),得到化合物1,7.20克白色固体,产率70.35%。Mp:162.1-162.7℃.IR(KBr)νmax(cm-1):1710,1610,1430,1290,802.1H NMR(500MHz,CDCl3)δ:8.00(d,J=8.2Hz,4H),7.36-7.26(m,2H),6.74(s,2H),6.26-6.14(m,2H),5.91(s,2H),5.55(s,1H),3.93(s,3H).
(2)5,15-二-(4-甲氧碳基苯基)-10,20-二苯基卟啉(化合物2)的合成:
取0.38克苯甲醛溶于20毫升三氯甲烷,氮气除氧20分钟后,室温下经恒压滴液漏斗避光滴加至100毫升含有0.95克化合物1的三氯甲烷溶液中,滴加1小时30分钟完毕,接着室温下避光反应4小时后,加入1.5克四氯苯醌反应过夜,再加入2毫升三乙胺,将反应混合物用200-300目硅胶柱层析粗提纯(淋洗液:三氯甲烷),得到黑色液体用无水硫酸钠干燥除水后,减压旋蒸后除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比3:1),得化合物2,0.16克紫色固体,产率12.89%。Mp:92.8-93.4℃.IR(KBr)νmax(cm-1):2960,1720,1600,1260,802.1H NMR(500MHz,CDCl3)δ:8.91-8.89(m,4H),8.87-8.80(m,4H),8.47(d,J=8.0Hz,4H),8.33(d,J=8.0Hz,4H),8.24(d,J=7.5Hz,4H),7.85-7.75(m,6H),4.14(s,6H),0.95-0.85(m,2H).
(3)5,15-二-(4-甲酸基苯基)-10,20-二苯基卟啉(化合物3)的合成:
取0.2克化合物2加入35毫升四氢呋喃和35毫升甲醇的混合溶液中,再加入10毫升含有1克氢氧化钾的水溶液,80℃冷凝回流反应10小时,冷却至室温后,用2mol/L的盐酸溶液酸化至pH为2~3之间,布氏漏斗抽滤,得化合物3,0.19克墨绿色固体,产率98.80%。Mp:>200℃.IR(KBr)νmax(cm-1):2960,1720,1600,1260,802.
(4)邻己烷氧基苯酚(化合物4)的合成:
取30克邻苯二酚、45克1-溴代正己烷、60克无水碳酸钾、2.28克碘化钾和300毫升无水乙醇,85℃冷凝回流反应12小时,冷却至室温后抽滤,滤液减压旋蒸除去溶剂,0.5mmHg的减压蒸馏情况下收集84℃的馏分,得化合物4,15.46克无色油状液体,产率29.52%。Bp:106±3℃.IR(KBr)νmax(cm-1):1260,930.1H NMR(300MHz,CDCl3)δ:6.95-6.82(m,4H),5.68(s,1H),4.02(t,J=6.6Hz,2H),1.83-1.76(m,2H),1.48-1.31(m,6H),0.91(t,J=6.9Hz,3H).
(5)邻二己烷氧基苯(化合物5)的合成:
取10克邻苯二酚、45克1-溴代正己烷、37.26克无水碳酸钾、3.32克碘化钾和125毫升无水乙醇,85℃冷凝回流反应60小时,冷却至室温后抽滤,滤液减压旋蒸除去溶剂,0.5mmHg的减压蒸馏情况下收集142℃的馏分,得化合物5,24.53克无色油状液体,产率96.96%。Bp:255±3℃.IR(KBr)νmax(cm-1):1250,939.1H NMR(300MHz,CDCl3)δ:6.89(s,4H),3.99(t,J=6.9Hz,4H),1.83-1.76(m,4H),1.49-1.31(m,12H),0.90(t,J=6.9Hz,6H).
(6)单羟基-五己烷氧基苯并菲(化合物6)的合成:
取1.16克化合物4和3.32克化合物5溶于60毫升二氯甲烷中,经恒压滴液漏斗滴加至80毫升含有12.96克无水三氯化铁和8毫升硝基甲烷的二氯甲烷溶液中,滴加30分钟完毕,控制反应液温度在0~3℃之间反应4小时后,加入30毫升甲醇和60毫升水终止反应,分出有机层,用30毫升二氯甲烷萃取水层三次,收集所有有机层,再用20毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/乙酸乙酯,体积比50:1),得到化合物6,1.80克白色固体,产率40.45%。Mp:47.7-50.1℃.IR(KBr)νmax(cm-1):3040,1240,837.1H NMR(400MHz,CDCl3)δ:7.96(s,1H),7.83(s,3H),7.82(s,1H),7.77(s,1H),5.91(s,1H),4.31-4.19(m,10H),1.98-1.90(m,10H),1.59-1.40(m,30H),0.96-0.92(m,15H)
(7)溴代十二氧基-五己氧基苯并菲(化合物7)的合成:
取1.19克化合物6、3.15克1,12-二溴十二烷、0.15克四丁基溴化铵、0.36克氢氧化钾、14毫升水和22毫升二氯甲烷,氮气氛围下室温搅拌反应24小时后,分出有机层,用15毫升二氯甲烷萃取水层三次,收集所有有机层,再用15毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/乙酸乙酯,体积比100:1),得到化合物7,1.20克乳白色固体,产率75.60%。Bp:>300℃.IR(KBr)νmax(cm-1):2930,1620,1260,836.1H NMR(400MHz,CDCl3)δ:7.84(s,6H),4.23(t,J=6.8Hz,12H),3.40(t,J=6.8Hz,2H),1.98-1.83(m,12H),1.67-1.50(m,16H),1.42-1.29(m,32H),0.93(t,J=6.8Hz,15H).
(8)苯并菲十二烷氧基桥连甲氧羰基苯基卟啉(化合物8)的合成:
取0.20克化合物3、0.73克化合物7、0.10克碳酸钾和15毫升N,N-二甲基甲酰胺溶液,氮气氛围下80℃回流反应32小时后,加入150毫升水,用75毫升乙酸乙酯萃取三次,分出水层和有机层,用20毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比1:1),得到化合物8,0.38克紫色固体,产率52.78%。Mp:25.6±3℃.IR(KBr)νmax(cm-1):2930,1720,1260.1H NMR(500MHz,CDCl3)δ:8.94-8.77(m,8H),8.46(d,J=8.0Hz,4H),8.32(d,J=8.0Hz,4H),8.24-8.22(m,4H),7.85-7.75(m,18H),4.58-4.47(m,4H),4.24(t,J=6.5Hz,24H),1.97-1.92(m,28H),1.66-1.39(m,92H),0.97-0.93(m,30H),-2.77(s,2H).Elemental analysis calcd for C166H218N4O16(2525):C,78.95,H,8.70,N,2.22,Found:C,77.68,H,8.65,N,2.43.
(9)苯并菲十二烷氧基桥连甲氧羰基苯基卟啉金属Zn配合物(化合物9)的合成:
取0.14克取化合物8、0.07克氯化锌、22毫升N,N-二甲基甲酰胺和15毫升三氯甲烷,氮气氛围下65℃回流反应2.5小时后,加入250毫升水,用20毫升三氯甲烷萃取三次,分出水层和有机层,用12毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/乙酸乙酯,体积比100:1),得到化合物9,0.12克紫色固体,产率84.39%。Mp:>300℃.IR(KBr)νmax(cm-1):2930,1720,1260.1H NMR(500MHz,CDCl3)δ:8.96-8.89(m,8H),8.46(d,J=8.0Hz,4H),8.33(d,J=8.0Hz,4H),8.23(d,J=8.0Hz,4H),7.80-7.67(m,18H),4.53(t,J=6.5Hz,4H),4.23-4.11(m,24H),1.97-1.88(m,28H),1.61-1.40(m,92H),0.98-0.94(m,30H).
- 还没有人留言评论。精彩留言会获得点赞!
- 沙格雷酯中间体2-[2-(3-甲氧基苯基)乙基]苯酚的合成方法
- 一种4,5,6-三氯-n-(2,3-二氟苯基)-2-(3-(三氟甲基)苯氧基)烟酰胺合成方法
- 一种甲基苯基乙烯基乙氧基硅烷的制备方法
- (2e, 6e)-2-(3,5-二甲氧基苯基亚甲基)-6-(4-氯苯基亚甲基)环己酮及其衍生物的新应用
- 一种基于3,4-二甲氧基苯基的取代苯甲酰胺新化合物、制备方法及用图
- 一种n-{[2,5-二乙氧基-4-[3-(3-甲氧基-苯基)-脲基甲基]-苯基}-甲磺酰胺新化合物 ...的制作方法
- 一种n-{[2,5-二乙氧基-4-[3-(4-甲氧基-苯基)-脲基甲基]-苯基}-甲磺酰胺新化合物 ...的制作方法
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 一种3-(2-硝基苯甲氧基)-1-(4-氯苯基)-1h-吡唑的绿色合成工艺的制作方法
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程