一种葡聚糖/吲哚美辛接枝物的合成方法与应用与流程
本发明涉及药用材料领域,具体涉及一种葡聚糖/吲哚美辛的合成以及用之制备成负载难溶性药物聚合物纳米粒溶液给药系统应用。
背景技术:
恶性肿瘤是现今危害人类健康、破坏人类生存质量的一大杀手,同时也是致死的主要原因之一,每年约有700多万人死于肿瘤(约占死亡总人数的13%);近年来,我国恶性肿瘤的发病率和死亡率也逐年上升,每年发病人数260多万,死亡达180万。目前临床上治疗肿瘤的方法包括手术切除,化学治疗,放射治疗等;其中应用药物进行化疗是手术切除治疗的的辅助手段之一,也是主要的全身治疗方法。但由于大多数药物水溶性很差,难以吸收,生物利用度很低,导致治疗效果不理想;包括加入增溶剂、助溶剂或使用有机溶剂等方法虽然能在一定程度上改善药物的溶解度,但同时也带来了很大的毒副作用。此外药物对组织器官的低选择性导致药物的毒副作用很大,常常有患者由于严重的毒副作用而被迫终止化疗,使药物的临床应用受到了很大的限制。因此,寻找新型的辅料,构建出合适的给药系统,以增大药物溶解度为出发点,制备出高效低毒靶向制剂是抗肿瘤药物研究的关键突破点。
聚合物纳米粒是在一个较长的亲水性骨架上接枝一个较小的疏水性基团,在溶剂中,它的疏水端聚集形成核芯,而亲水端则形成外层;在药剂学中,纳米粒具有特殊的医疗价值,将药物较为稳定地包裹于核芯中,则能够暂时隐藏药物的性质,它在体内的过程和生物相容性将取决于亲水外层的性质。因此我们又可以通过对亲水、疏水基团的改造和修饰,赋予其特有的性质,将抗癌药物带入靶器官甚至于肿瘤细胞,设计出高效低毒的靶向制剂。此前,本实验室已制备过以壳寡糖作为亲水骨架,吲哚美辛作为疏水链段的两亲性纳米粒,研究壳寡糖/吲哚美辛接枝物作为抗肿瘤药物载体的可能性,并取得了一定的成果。但壳寡糖上大量游离氨基的存在,使得由壳寡糖制成的纳米粒在水中一般带有较高的正电荷,它在水中的结构类似于阳离子表面活性剂,毒性较大。而葡聚糖是迄今为止被美国fda批准的临床可直接用于血液制品的高分子材料。具有非常好的生物相容性和安全性。因此选择何种物质作为聚合物纳米粒的亲水基团和疏水基团是关系到最终制剂的药效和安全性的重要因素,也是一切工作的前提。
技术实现要素:
为解决有些药物存在严重毒副作用的问题,本发明提出一种葡聚糖/吲哚美辛接枝物的合成方法与应用,以葡聚糖/吲哚美辛接枝物得到的给药系统具有高载药量和包封率,呈低毒性,可作为药物载体使用。
本发明是通过以下技术方案实现的:以4-二甲氨基吡啶(dmap)为催化剂,碳二亚胺(edc)为脱水剂,以葡聚糖和吲哚美辛分别作为亲水骨架和疏水链段,通过使葡聚糖(dex)化学结构中的羟基与吲哚美辛(indo)化学结构中的羧基发生酯化反应,生成两亲性化合物葡聚糖/吲哚美辛接枝物,方法合成为:
分别称取吲哚美辛(indo)、碳二亚胺和4-二甲氨基吡啶,溶于二甲基亚砜(dmso)中,搅拌使其溶解后,恒温15-80℃保持20min-24h,然后加入葡聚糖(dex),15-80℃搅拌反应2-72h后,将反应液转移至透析袋中,以双蒸水为介质进行透析,持续更换介质4-96h,透析完毕后,收集透析袋内的液体,离心后取上清液冷冻干燥即得葡聚糖/吲哚美辛接枝物冻干品(缩写为dex/indo)。
反应结构式如下所示:
作为优选,吲哚美辛、碳二亚胺、4-二甲氨基吡啶的摩尔比为1∶1-10∶0.1-1。
作为优选,所述葡聚糖与吲哚美辛的摩尔比为1∶0.05-2,葡聚糖的分子量为2500-100000。通过改变吲哚美辛的不同投料量,合成一系列理论接枝率在1-100%之间的葡聚糖/吲哚美辛接枝物(dexmw/indo理论接枝比例)。这些载体溶于水后可形成粒径在10-200nm,ζ电位在-10-10mv,临界聚集浓度为150-1000μg/ml的微粒给药系统,该空白载体的hepg2细胞毒性(ic50)在50-2000μg/ml之间。
作为优选,透析袋的截留分子量为葡聚糖分子量的1/3-3/4。
作为优选,离心条件为2-10℃,1500-10000rpm离心时间5-45min。
葡聚糖又称右旋糖酐,易溶于水,理化性质稳定,毒性低,是一种较为理想的亲水材料,葡聚糖毒性很低,血液相容性较好,同时葡聚糖的碳链上含有大量游离的自由羟基,为接枝疏水性基团或其他功能基团提供了可能。而聚合物纳米粒的疏水基团,则担负着包裹难溶性药物的任务;疏水基团的化学组成、空间结构和数量,决定着载体的载药量,直接影响到药物的药效;同样具有大π键的化合物,吲哚美辛作为纳米粒的疏水基团,它们的π键能与难溶性药物相互作用形成共轭,增加了与难溶性药物之间的作用力,从而提高了难溶性药物在水中的溶解度,也增加了难溶性药物进入水溶性介质后的稳定性;而葡聚糖表面电位很低,几乎等于0,葡聚糖的生物相容性更高。
本发明所述的所述的一种葡聚糖/吲哚美辛接枝物的合成方法得到的葡聚糖/吲哚美辛接枝物在提供负载难容性药物的葡聚糖/吲哚美辛的纳米粒溶液给药系统上的应用。
将葡聚糖/吲哚美辛接枝物溶解于二甲基亚砜中,得到接枝物溶液,然后称取难溶性药物,溶解于二甲基亚砜,得到难溶性药物溶液,在磁力搅拌下,将难溶性药物溶液滴入接枝物溶液中,先冰浴探头超声,再室温避光搅拌4-24h后,将反应液转移至透析袋中,以双蒸水为介质进行透析以除去二甲基亚砜,持续更换介质4-96h后,将袋内液体离心后取上清液,即得负载难溶性药物的葡聚糖/吲哚美辛的纳米粒溶液。
载药纳米粒溶液既可制成冻干粉末,也可制成口服给药系统。
所述接枝物溶液溶液的浓度为0.1-5mg/ml,难溶性药物溶液的浓度为0.05-10mg/ml,葡聚糖/吲哚美辛接枝物与难溶性药物的理论质量比为1∶0.05-2。
难溶性药物选自碱化阿霉素、紫杉醇、羟基喜树碱、吲哚美辛、环孢素a、甲氧苄啶,卡马西平,磺胺甲噁唑,茶碱、乙胺嘧啶、顺铂、二甲硅油、二甲磺酸阿米三嗪、二氟尼柳、二硫化硒、十一酸睾酮、三硅酸美、己烯雌酚、己酸羟孕酮、马来酸伊索拉定、厄贝沙坦、比沙可定、卡莫司汀、氟尿嘧啶、巯嘌呤、甲氨蝶呤、放线菌素d、阿莫西林、头孢氨苄、氯霉素、利福平、磺胺嘧啶、甲氧卞啶、阿昔洛韦、齐多夫定、阿苯达唑、吡喹酮、硫酸奎尼、青蒿素中一种。
冰浴探头超声为100-800w,超声1-4s,间隔时间大于超声时间,共15-80次。
作为优选,透析袋的截留分子量为葡聚糖分子量的1/3-3/4。
作为优选,将透析袋内液体置于容量瓶中,用双蒸水定容至刻度,在离心条件为2-15℃,1500-10000rpm离心时间5-45min,取上清液。
本发明所制备得到的载有难溶性药物、以葡聚糖/吲哚美辛为空白载体的微粒给药系统。该载药系统在水溶液中可形成粒径为50~1000nm、zeta电位为-30mv~+30mv的微粒。微粒的载药量为1%~80%,包封率为50%~90%。药物在体外漏槽条件下表现出缓慢释放特征,起到控释功能,该微粒给药系统的肿瘤细胞抑制率也得到提高。
与现有技术相比,本发明的有益效果是:
(1)本发明中的葡聚糖/吲哚美辛,葡聚糖为酶解的低分子,完全水溶,吲哚美辛含有和难溶性药物相似的大π键的结构,和药物之间存在着范德华力,因此,制备得到的给药系统具有高载药量和包封率;
(2)通过选用不同分子量的葡聚糖和不同接枝率的吲哚美辛,可以达到对药物的控、缓释放。葡聚糖/吲哚美辛空白纳米粒在细胞上呈现低毒性,可以作为药物载体使用;(3)本发明所述的葡聚糖/吲哚美辛微粒给药系统,制备简单,无有机溶剂残留,对肿瘤细胞的抑制率强,并具有肝、肾、肿瘤靶向性,尤其在心、肺聚集较少。
附图说明
图1为葡聚糖的核磁共振氢谱;
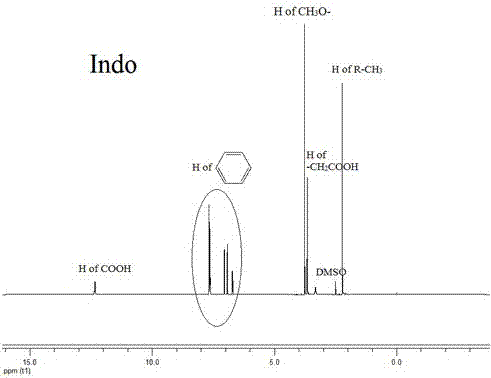
图2为吲哚美辛的核磁共振氢谱;
图3为葡聚糖/吲哚美辛接枝物的核磁共振氢谱;
图4为实施例1制备的dex10000/indo20%接枝物纳米粒在双蒸水中的临界聚集浓度图;
图5为实施例2制备的dex10000/indo50%接枝物纳米粒在双蒸水中的临界聚集浓度图;
图6为实施例3制备的dex10000/indo100%接枝物纳米粒在双蒸水中的临界聚集浓度图;
图7为实施例1制备的dex10000/indo20%接枝物纳米粒透射电子显微镜图;
图8为实施例2制备的dex10000/indo50%接枝物纳米粒透射电子显微镜图;
图9为实施例3制备的dex10000/indo100%接枝物纳米粒透射电子显微镜图;
图10为dex10000/indo100%-dox10%的透射电子显微镜图;
图11为dex10000/indo20%-dox10%载药纳米粒在不同ph值的介质中的释放曲线:
图12为dex10000/indo50%-dox10%载药纳米粒在不同ph值的介质中的释放曲线:
图13为dex10000/indo100%-dox10%载药纳米粒在不同ph值的介质中的释放曲线:
图14为dex10000/indo20%对hep-g2细胞的生长抑制曲线;
图15为dox·hcl对hep-g2生长抑制图;
图16为dex10000/indo20%-dox10%对hep-g2生长抑制图;
图17为balb/c小鼠注射药物6、24、48h后,心脏组织切片的荧光照片;
图18为balb/c小鼠注射药物6、24、48h后,肝脏组织切片的荧光照片;
图19为balb/c小鼠注射药物6、24、48h后,脾脏组织切片的荧光照片;
图20为balb/c小鼠注射药物6、24、48h后,肺组织切片的荧光照片;
图21为balb/c小鼠注射药物6、24、48h后,肾脏组织切片的荧光照片;
图22为dox·hcl和dau·hcl高效液相色谱图;
其中:1-dox·hcl,2-dau·hcl;
图23为各脏器匀浆液的hplc图;
其中:(1)为空白匀浆液经前处理后的hplc图;(2)为含有dox·hcl和dau·hcl的匀浆液经前处理后的hplc图;(a)心脏(b)肝脏(c)脾脏(d)肺(e)肾脏;
图24为dox·hcl在balb/c小鼠体内各脏器的分布情况;
图25为载阿霉素纳米粒在balb/c小鼠体内各脏器的分布情况;
具体实施方式
下面通过实施例对本发明作进一步详细说明,实施例中所用原料均可市购或采用常规方法制备。
实施例1
分别称取的吲哚美辛(3.086mmol,1103mg),edc(9.258mmol,1774mg)和dmap(0.9258mmol,113.0mg),溶于10mldmso,搅拌使其完全溶解后,恒温60℃保持30min,缓慢加入葡聚糖(3.086mmol,500mg)(mw=10000),25℃搅拌反应24h后,将反应液转移至透析袋(mwco=7000)中,以双蒸水为介质进行透析,持续更换介质48h,透析完毕后,收集透析袋内的乳状液体,于4℃,4000rpm离心10min,除去不溶性物质,取上清液冷冻干燥即得葡聚糖/吲哚美辛接枝物(以下缩写为dex/indo)冻干品1。
所得的葡聚糖/吲哚美辛接枝物冻干品1为dex10000/indo20%。
实施例2
分别称取的吲哚美辛(1.543mmol,551.5mg),edc(4.629mmol,886.9mg)和dmap(0.4629mmol,56.0mg),溶于10mldmso,搅拌使其完全溶解后,恒温70℃保持20min,缓慢加入葡聚糖(3.086mmol,500mg)(mw=10000),70℃搅拌反应12h后,将反应液转移至透析袋(mwco=5000)中,以双蒸水为介质进行透析,持续更换介质48h,透析完毕后,收集透析袋内的乳状液体,于10℃,2000rpm离心50min,除去不溶性物质,取上清液冷冻干燥即得葡聚糖/吲哚美辛接枝物(以下缩写为dex/indo)冻干品2。
所得的葡聚糖/吲哚美辛接枝物冻干品2为dex10000/indo50%。
实施例3
分别称取的吲哚美辛(0.617mmol,220.6mg),edc(1.851mmol,354.7mg)和dmap(0.1851mmol,23.0mg),溶于10mldmso,搅拌使其完全溶解后,恒温20℃保持12h,缓慢加入葡聚糖(3.086mmol,500mg)(mw=10000),20℃搅拌反应48h后,将反应液转移至透析袋(mwco=6000)中,以双蒸水为介质进行透析,持续更换介质48h,透析完毕后,收集透析袋内的乳状液体,于2℃,8000rpm离心20min,除去不溶性物质,取上清液冷冻干燥即得葡聚糖/吲哚美辛接枝物(以下缩写为dex/indo)冻干品3。
所得的葡聚糖/吲哚美辛接枝物冻干品3为dex10000/indo100%。
测试例1:葡聚糖/吲哚美辛接枝物的结构确证
采用核磁共振氢谱(1hnmr)对葡聚糖/吲哚美辛接枝物的结构进行确证,称取dex10000和indo各10mg,分别溶于0.5ml氘代dmso,使其最终浓度分别都为20mg/ml;称取三种接枝物各20mg,分别溶于0.5ml氘代dmso中,使其最终浓度为40mg/ml,用核磁共振仪分别记录其氢谱,通过比对接枝物与葡聚糖及吲哚美辛的氢谱,如图1、图2、图3所示。
对它们的化学结构进行分析:图1中有葡聚糖的-ch2-质子信号(化学位移约为3.4ppm)和-ocho-的质子信号(化学位移约为5.0ppm);图2中有吲哚美辛中苯环上的质子信号(化学位移在6~8ppm)以及羧基上的质子信号(化学位移约为12.3ppm);图3中,我们不仅检测到了葡聚糖中的亚甲基质子信号(化学位移约为3.4ppm),也检测到了吲哚美辛中苯环上的质子信号(化学位移在6~8ppm),但没有检测到吲哚美辛中羧基上的质子信号(化学位移约为12.3ppm),说明通过酯化反应,吲哚美辛已经被成功接枝到葡聚糖的骨架上。
测试例2:葡聚糖/吲哚美辛接枝物的理化性质研究
葡聚糖/吲哚美辛接枝物具有一端亲水,一端疏水的两亲性结构;当它在水中达到一定的浓度之后,接枝物分子相互聚集,形成亲水基向外,疏水基向内的缔合体,即胶束结构;此分子聚集形成胶束结构的最低浓度即为其临界聚集浓度(criticalaggregationconcentration,cac);采用芘荧光法测定纳米粒在水中的cac值。实验步骤如下:
(1)芘的丙酮溶液的配制
称取荧光探针物质芘3mg,加入适量丙酮使其全溶后,转移至50ml容量瓶中,用丙酮稀释至刻度,摇匀即得60μg/ml芘的丙酮溶液;移取此溶液1ml于50ml容量瓶中,用丙酮定容即得1.2μg/ml芘的丙酮溶液。
(2)纳米粒溶液的配制
称取葡聚糖/吲哚美辛接枝物50mg,分散于10ml双蒸水中,探头冰浴超声(20次,400w,工作2s,停止4s)后转移至25ml容量瓶中,用双蒸水定容,得到2mg/ml的纳米粒溶液。用双蒸水将此纳米粒溶液分别稀释成浓度为4~2000μg/ml的系列溶液。
(3)芘荧光法测定纳米粒的cac
分别移取0.5ml上述1.2mg/ml芘的丙酮溶液于15支试管中,避光挥干丙酮;向试管中分别加入5ml上述1.2项下配制的纳米粒系列溶液,将此系列溶液于37℃,100rpm的水浴振荡器中,恒温振荡24h,使芘在溶液中的最终浓度为1.2×10-7g/ml;设定荧光激发波长为339nm,扫描系列溶液在350~450nm的荧光发射光谱,以第一个吸收峰(i1=373nm)和第三个吸收峰(i3=391nm)吸光度的比值变化来确定cac,结果如图4、图5、图6所示。
测试例3:纳米粒粒径及表面电位的测定
分别称取实施例1-3的dex/indo接枝物10mg,分散于5ml双蒸水中,探头冰浴超声(20次,400w,工作2s,停止4s后,用双蒸水定容至10ml,得到1mg/ml的纳米粒溶液,用微粒粒度与表面电位测定仪测定此纳米粒溶液的粒径和表面电位,结果见表1所示;
表1:dex/indo纳米粒的性质
从上述数据中可以看出,随着接枝率的增大,纳米粒的临界聚集浓度逐渐变小,粒径逐渐减小,而表面电位呈现出增大的趋势。
测试例4:透射电子显微镜(tem)观察接枝物的形态
tem是用于观测微粒粒子大小、形态和分散情况的常用工具,分别称取实施例1-实施例3制备的dex/indo接枝物5mg,分散于5ml双蒸水中,探头冰浴超声(20次,400w,工作2s,停止4s)后,用双蒸水定容至25ml,得到0.2mg/ml的溶液;分别移取1滴上述纳米粒溶液于覆盖有碳膜的电镜铜网上,1%醋酸双氧铀染色,吸弃表面多余的液体,风干后在透射电镜(tem)下观察葡聚糖/吲哚美辛接枝物的形态,放大倍数为200,000倍,标尺为100nm,结果如图7、图8、图9所示。
应用例
阿霉素的碱化方法详述如下:称取适量盐酸阿霉素溶于二甲基亚砜(dmso)中,使质量浓度为10g/l,加入2倍摩尔量的三乙胺,避光搅拌过夜使脱盐酸。然后将反应液放入透析袋中(mwco=1000),避光透析,透析液为去离子水,开始间隔半小时换一次水,换5次后间隔1小时换一次水,共透析12h,最后冷冻干燥即得碱化阿霉素(dox)的冻干粉末。
应用例1载阿霉素葡聚糖/吲哚美辛纳米粒的制备
称取实施例1接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物溶液的浓度为2mg/ml。称取浓度为1mg/mldox的dmso溶液1ml在磁力搅拌下缓慢滴入接枝物溶液中,冰浴探头超声(400w,超声2s,间隔4s,40次),室温避光搅拌12h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=7000),以双蒸水为介质进行透析以除去dmso。持续更换介质24h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载碱性阿霉素葡聚糖/吲哚美辛的dex10000/indo20%-dox10%纳米粒溶液。
应用例2
称取实施例2接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取浓度为1mg/mldox的dmso溶液1ml,在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(400w,超声2s,间隔4s,40次),室温避光搅拌12h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=7000),以双蒸水为介质进行透析以除去dmso。持续更换介质24h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载碱性阿霉素葡聚糖/吲哚美辛的dex10000/indo50%-dox10%纳米粒溶液。
应用例3
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取浓度为1mg/mldox的dmso溶液0.5ml,在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(400w,超声2s,间隔4s,40次),室温避光搅拌12h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=7000),以双蒸水为介质进行透析以除去dmso。持续更换介质24h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载碱性阿霉素葡聚糖/吲哚美辛的dex10000/indo100%-dox5%纳米粒溶液。
应用例4
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取浓度为1mg/mldox的dmso溶液1ml,在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(800w,超声1s,间隔2s,60次),室温避光搅拌20h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=6000),以双蒸水为介质进行透析以除去dmso。持续更换介质48h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于8℃,5000rpm离心20min,取上清液,即得负载碱性阿霉素葡聚糖/吲哚美辛的dex10000/indo100%-dox10%纳米粒溶液。
应用例5
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取浓度为1mg/mldox的dmso溶液1.5ml,在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(200w,超声4s,间隔6s,70次),室温避光搅拌5h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=6000),以双蒸水为介质进行透析以除去dmso。持续更换介质72h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载碱性阿霉素葡聚糖/吲哚美辛的dex10000/indo100%-d0x15%纳米粒溶液。
应用例6
称取实施例2接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取浓度为1mg/ml的环孢素a/乙醇溶液1ml,在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(100w,超声3s,间隔4s,20次),室温避光搅拌8h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=5000),以双蒸水为介质进行透析以除去dmso、乙醇。持续更换介质72h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于10℃,2000rpm离心40min,取上清液,即得负载环孢素a葡聚糖/吲哚美辛的纳米粒溶液。
应用例7
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。按照紫杉醇/接枝物的理论投料比(w/w),称取10mg/ml紫杉醇/乙醇溶液1ml。在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(500w,超声2s,间隔3s,50次),室温避光搅拌20h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=4000),以双蒸水为介质进行透析以除去dmso、乙醇。持续更换介质10h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于7℃,7000rpm离心30min,取上清液,即得负载紫杉醇葡聚糖/吲哚美辛的纳米粒溶液。
应用例8
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。按照药物:接枝物的理论投料比(w/w),称取1mg/ml卡马西平的三氯甲烷溶液1ml。在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(400w,超声2s,间隔4s,40次),室温避光搅拌12h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=7000),以双蒸水为介质进行透析以除去dmso、三氯甲烷。持续更换介质24h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载卡马西平葡聚糖/吲哚美辛的纳米粒溶液。
应用例9
称取实施例3接枝物10mg,溶于约3mldmso,待其完全溶解后,定容至5ml,使接枝物的浓度为2mg/ml。称取配制成1mg/ml青蒿素的丙酮溶液1ml。在磁力搅拌下将此溶液缓慢滴入接枝物溶液中,冰浴探头超声(400w,超声2s,间隔4s,40次),室温避光搅拌12h,使药物与载体充分混合后,将反应液转移至透析袋中(mwco=7000),以双蒸水为介质进行透析以除去dmso、丙酮。持续更换介质24h后,将袋内液体置于25ml容量瓶中,用双蒸水定容至刻度,将产物于4℃,3000rpm离心15min,取上清液,即得负载青蒿素葡聚糖/吲哚美辛的纳米粒溶液。
测试应用例1:载阿霉素葡聚糖/吲哚美辛纳米粒载药量和包封率的测定
分别移取应用例1-应用例5制备的载阿霉素纳米粒50μl于5ml容量瓶中,用dmso稀释至刻度,水浴超声30min后,荧光测定吸光度,以同浓度的空白纳米粒作为对照,计算药物浓度,由下式计算包封率(encapsulationefficiency,ee)和载药量(drug-loadingrate,dl),结果如表2所示。
其中c’为实测药物总浓度,v为载药纳米粒溶液体积,wd为实际投药量,wc为载药纳米粒溶液中载体的质量
表2:载药纳米粒的载药量和包封率
测试应用例2:载阿霉素葡聚糖/吲哚美辛纳米粒粒径及表面电位的测定
以微粒粒度与表面电位分析仪测定载药纳米粒分散于双蒸水中的粒径和表面电位,结果如表3所示。
表3载药纳米粒的粒径与表面电位
从表3中可知,相比于空白纳米粒,载药纳米粒的粒径均增大,表面电位均有减小的趋势。
测试应用例3:tem观察载药纳米粒的形态
移取1滴dex10000/indo100%-dox10%纳米粒溶液于覆盖有碳膜的电镜铜网上,1%醋酸双氧铀染色,吸弃表面多余的液体,风干后在透射电镜(tem)下观察载dox纳米粒的形态,结果如图10所示,放大倍数为20,0000倍,标尺为100nm。.
从图10中看出,载药纳米粒粒子呈类球形,分散良好;对比图7,载药纳米粒的分散更为均匀,基本无团聚现象。
测试应用例4:载药纳米粒的体外释放行为研究
采用透析袋法考察载药纳米粒在37℃,不同ph值(ph=5.0,6.8,7.4)磷酸缓冲液中的体外释放行为:取已知载药量的载药纳米粒溶液,用双蒸水调整其浓度为(以dox计)50μg/ml,取此溶液1ml于透析袋中,将透析袋分别置于5ml上述缓冲液中;在37℃,100rpm的水浴恒温振荡器中,分别在1,2,4,6,8,12,24,36,48,60,72h取样测定,为了保证达到漏槽条件,每次取样时均取出全部介质,并补充5ml新鲜介质于其中;取样后荧光测定吸光度,计算药物的累积释放率,同法平行测定三组,结果如图11、图12、图13所示。
由释放曲线可以看出,随着ph值的增加,dox的释放速率有增大的趋势,它在酸性介质中释放较快,而在碱性介质中释放较慢;随着吲哚美辛接枝率的增加,dox越来越难从疏水核芯中释放出来,最慢的在72h内释放不足10%。
对比例1:葡聚糖/吲哚美辛纳米粒及载阿霉素纳米粒的细胞评价
(1)空白纳米粒溶液在hep-g2细胞上的毒性考察
按设定的纳米粒的系列浓度,以每孔总体积为200μl,计算出各孔所需的纳米粒质量。
取出已贴附有hep-g2细胞的96孔培养板,吸弃板中原培养液,用pbs溶液清洗细胞后,按各孔所需的纳米粒质量加入母液,控制每孔加入体积均在50μl以内;补加dmem培养液至200μl,使各孔中载体的最终浓度为设定的系列浓度,每个浓度设三个复孔,置于培养箱中培养育72h。
72h后取出培养箱中的96孔板,向各孔中分别加入20μlmtt溶液,于培养箱中反应4h后,吸弃上清液,向各孔中加入150μldmso,振摇10min后,用酶联免疫检测仪测定各孔在570nm处的吸光度。按下式计算细胞生长抑制率:
其中a570(treated)为实验组的吸光度,a570(control)为空白对照组的吸光度。
采用改良寇式法计算ic50值:
xm:lg(最大剂量);i:lg(最大剂量/相邻剂量);p:抑制率之和
pm:最大抑制率;pn:最小抑制率
以未加载体的细胞为空白对照,同法操作。
dex10000/indo20%对hep-g2细胞的生长抑制曲线如图14所示,实施例1-实施例3纳米粒对hep-g2细胞的ic50值如表4所示。
表4空白纳米粒对hep-g2的ic50值
由表4得出,随着吲哚美辛接枝率的增大,纳米粒的细胞毒性越来越大。
(2)载药纳米粒对hep-g2细胞的药效学考察
以dox·hcl作为对照,使得游离的阿霉素溶液中的阿霉素质量与dex10000/indo20%-dox纳米粒中测得的实际阿霉素量相同。实验方法与上文中空白纳米粒对hep-g2细胞的毒性考察相同,考察载药纳米粒对hep-g2细胞增殖的抑制作用。
dox·hcl和载阿霉素纳米粒对细胞的ic50值及生长的抑制情况分别如表5所示和图15、图16所示。
表5dox·hcl和dex10000/indo20%-dox对hep-g2的ic50值
载药纳米粒的ic50值小于dox·hcl,差异有统计学意义(p<0.01),显示出良好的抗肿瘤效果。
对比例2:载阿霉素葡聚糖/吲哚美辛纳米粒体内分布的初步研究
(1)荧光观察法考察纳米粒在小鼠体内各脏器的分布情况
取6-8周龄balb/c小鼠12只,分为四组,每组3只,分别以6mg/kg的剂量尾静脉注射dox·hcl和dex/indo-dox(剂量以dox计),分别以50mg/kg的剂量尾静脉注射fitc-dex/indo和fitc-dex/indo-dox,各组分别于6h,24h,48h各颈椎脱臼处死一只,取出心、肝、脾、肺、肾组织,用生理盐水棉球擦拭血迹后使用oct包埋固定,保存于-80℃,使用冰冻切片机切成厚度为20μm的切片,置于荧光显微镜下观察,结果如图17~图21所示;其中,fitc呈现绿色荧光,dox及dox·hcl呈现红色荧光。(2)hplc法测定阿霉素在小鼠体内的分布情况
1、样品取材及预处理
取6-8周龄balb/c小鼠48只,分为两组,每组24只;一组按6mg/kg的剂量注射dox·hcl,另一组按6mg/kg(以dox计)的剂量注射dex10000/indo20%-dox,分别于12h,24h,48h,72h(每个时间点各6只)颈椎脱臼处死,取出心、肝、脾、肺、肾组织,用生理盐水棉球擦拭血迹后待处理。样品的前处理步骤如下:
心、肺组织的预处理:取小鼠心(或肺)组织称重,剪碎,分散于适量生理盐水中,置于研磨器中研磨均匀后,用生理盐水定容配成5%(w/v)的匀浆液,分别取0.15ml上述匀浆液于5ml离心管中,加入100μl2μg/ml的dau·hcl(盐酸柔红霉素)内标液,涡旋分散3min使药物均匀分布于组织匀浆液中,加入1ml提取溶剂,旋涡振荡3min后,于12000rpm离心3min,取下层的有机相用氮气吹干,残渣溶于1ml流动相后过0.45μm微孔滤膜进样。
肝脏、肾、脾脏组织的预处理:取小鼠肝脏(或肾、脾脏)组织称重,剪碎,分散于适量生理盐水中,置于研磨器中研磨均匀后,用生理盐水定容配成10%(w/v)的匀浆液,分别取0.15ml上述匀浆液于5ml离心管中,加入100μl4μg/ml的dau·hcl内标液,涡旋分散3min使药物均匀分布于组织匀浆液中,加入1ml提取溶剂,旋涡振荡3min后,12000rpm离心3min,取下层的有机相用氮气吹干,残渣溶于1ml流动相后过0.45μm微孔滤膜进样。
2、色谱条件的确定
高效液相色谱仪-荧光检测器;色谱柱:依利特hypersilods2,5μm,4.6mm×250mm;流动相∶乙腈-ph2.5磷酸缓冲液(35∶65),流速:1ml/min,柱温:25℃,检测波长ex=467nm,em=589nm,进样量:20μl
在上述色谱条件下,dox·hcl和dau·hcl的分离度符合要求如图22所示,色谱峰良好。
3、分别取小鼠心、肝、脾、肺、肾空白匀浆液0.15ml,不加任何药物,按3项下处理进样;分别取小鼠心、肝、脾、肺、肾空白匀浆液0.15ml,分别加入100μldox·hcl工作液(心、肺为2μg/ml的溶液,肝、肾、脾为4μg/ml的溶液),按3项下加入dau·hcl内标液,处理进样;两者对比可见组织中内源性物质对测定基本无干扰,分离度良好,色谱图如图23所示。
4、线性关系的考察
心、肺组织标准曲线的制备:分别取0.05,0.125,0.25,0.5,1.25,2.5,5,10ml上述2μg/ml的dox·hcl溶液于10ml容量瓶中,用双蒸水稀释至刻度,即得浓度为10~2000ng/ml的dox·hcl系列溶液。分别取0.15ml上述5%的匀浆液(w/v)于8只5ml离心管中,加入100μl2μg/ml的dau·hcl内标液,分别加入100μl浓度为10~2000ng/ml的dox·hcl系列溶液;涡旋3min使药物均匀分布于组织匀浆中,加入1ml提取溶剂,旋涡振荡3min后,12000rpm离心3min,取有机相用氮气吹干,残渣溶于1ml流动相后过0.45μm微孔滤膜进样。
肝脏、肾、脾脏组织标准曲线的制备:分别取0.05,0.125,0.25,0.5,1.25,2.5,5,10ml上述4μg/ml的dox·hcl溶液于10ml容量瓶中,用双蒸水稀释至刻度,即得浓度为20~4000ng/ml的dox·hcl系列溶液。分别取0.15ml上述10%的匀浆液(w/v)于8只5ml离心管中,加入100μl4μg/ml的dau·hcl内标液,分别加入100μl浓度为20~4000ng/ml的dox·hcl系列溶液;涡旋3min使药物均匀分布于组织匀浆中,加入1ml提取溶剂,旋涡振荡3min后,12000rpm离心3min,取有机相用氮气吹干,残渣溶于1ml流动相后过0.45μm微孔滤膜进样。
以dox·hcl在样品中的浓度为横坐标,dox·hcl与dau·hcl的峰面积比为纵坐标进行线性回归,并计算药物在各组织器官内的检测范围,方程如表6所示,证明此方法应用于各脏器中线性关系良好。精密度、回收率和稳定性均符合要求。
表6:线性关系的考察
5、hplc体内分析结果
药物在balb/c小鼠体内各脏器的分布如表6和图24所示:
表6:药物在balb/c小鼠体内各脏器中的浓度(μg/g)
hplc实验进一步证明了荧光观察的结果,dox·hcl组的各个脏器均在6h时浓度最大,之后就逐渐降低,而载药纳米粒组的各脏器基本在48h左右达到浓度的最大值,证明了载药纳米粒在体内的缓释作用。纵观四个时间点,载药纳米粒组中药物在心脏中的浓度均小于dox·hcl组,约在1/6~1/3之间,为解决阿霉素的心脏毒性提供了参考;除6h外,载药纳米粒组中药物在肝脏中的浓度均大小于dox·hcl组,最高为8倍,证明药物具有肝靶向性。药物在脾脏中具有一定量的的分布,尤其在48h时达到了17.97μg/g,说明药物对脾脏也有一定的靶向性;dox·hcl组中,在6h~72h的时间内,药物在肾脏中的浓度逐渐减小,而载药纳米粒组的药物在48h时达到最大,说明药物通过肾脏代谢。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种HDPE多单体接枝聚合物及其制备方法与制造工艺
- 接枝用的夹具的制作方法
- 一种两嵌段低分子聚合物接枝纳米二氧化硅粒子、制备方法和聚酯的制作方法
- 基于聚醚接枝改性的热增黏聚合物及其制备方法
- 以马铃薯淀粉为原料一锅法接枝聚合合成含钾高吸水树脂的方法
- 以马铃薯淀粉为原料氧化、碱糊化及接枝聚合一锅法制备吸附Ni<sup>2+</sup>离子树脂的方法
- 以马铃薯淀粉为原料氧化、碱糊化及接枝聚合一锅法制备吸附Co<sup>2+</sup>离子树脂的方法
- 以马铃薯淀粉为原料氧化、碱糊化及接枝聚合一锅法制备吸附Ag<sup>+</sup>离子树脂的方法
- 一种合成温敏含糖聚合物接枝的纳米金胶束的方法
- 一种基于点击化学的超支化聚合物接枝碳纳米管的制备方法
- 星形聚合物接枝多肽和聚乙烯亚胺的基因载体及制备方法与流程
- 一种木质纤维接枝丙烯酰胺聚合物及其制备方法与流程
- 使用新颖的阳离子性接枝聚合物的目标物分离浓缩方法与流程
- 含橡胶接枝聚合物粉体、以及含有其的太阳能电池用密封材料和夹层玻璃用中间膜的制作方法与工艺
- 一种原位制备黑磷混杂聚合物的制备方法与流程
- 附接到可降解的聚合物区的支撑件的制作方法与工艺
- 接枝聚合物、树脂着色物、该树脂着色物的制造方法和包含该树脂着色物的树脂组合物与流程
- 接枝聚合物、树脂着色物、该树脂着色物的制造方法和包含该树脂着色物的树脂组合物与流程
- 角蛋白接枝水性聚氨酯聚合物皮革复鞣填充材料的制备方法与流程
- 一种高接枝率聚烯烃接枝物的制备方法及该高接枝率聚烯烃接枝物的应用与流程