一种用于对不同樟树进行基因分型的SNP引物及检测方法与流程
技术领域:
:本发明属于生物
技术领域:
,具体涉及一种用于对不同樟树进行基因分型的snp引物及检测方法。
背景技术:
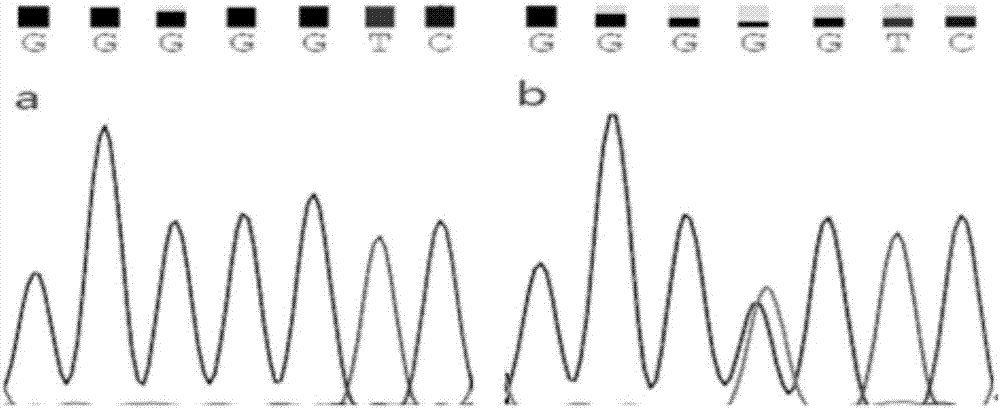
:樟树,别名香樟、芳樟、小叶樟等,隶属于樟科(lauraceae)樟属(cinnamomum),在我国具有重要的经济价值,且广泛分布于我国亚热带地区,如海南、江西、湖南、浙江、重庆、等省市区。樟树具有树姿雄伟、四季浓绿、灭菌驱虫和挥发香气等特点,发挥着重要的材用、药用、香料、油用和生态环境建设等价值。樟树因其可以提取天然樟脑和芳香油,而这两者分别是重要的工业和医药原料,因此针对樟树的研究也颇受关注。单核苷酸多态性(singlenucleotidepolymorphisms,snp)主要是指由于单个核苷酸的变异而引起基因组水平上的dna序列多态性,形式包括单碱基的缺失、插入、转换及颠换等。snp是一种最常见的遗传变异,具有遗传稳定性高、位点丰富且分布广泛、富有代表性、检测快速、易于实现自动化分析等特点。另外,由于snp自身具有密度高、分布广等特点,又加上先进的高通量snp检测系统的研制,snp标记将对未来生命科学领域产生重大的影响。hrm(high-resolutionmelting)分析技术是应用于snp检测分析的一项新技术,是由犹他大学和idaho科技公司合作开发的,该技术主要利用不同双链dna分子的片段长短、gc含量和分布不同,在加热变性时形成随温度变化的特定熔解曲线,进而利用饱和荧光染料和高精密度采集荧光信号仪器进行检测。基于hrm技术的原理,hrm检测不需要使用针对目标位点的特异性等位引物或探针,只需一对引物就可以对等位基因的变化进行识别。其灵敏度高、特异性强、简单快速、高效、经济,还具有高通量、闭管操作等优点,非常经济有效。因此,hrm大大简化了snp检测的操作步骤,并减少了分析时间,目前该项技术主要应用于突变扫描、snp的发现、基因分型、小片段的插入和删除、甲基化研究及混合样品的突变发现等方面。樟树生化类型准确划分,是高效开发有着重要的经济价值的樟树精油的基础,快速、准确的基因分型对化学类型的区分至关重要,目前关于此类研究,鲜见报道。技术实现要素:发明目的:针对现有技术中存在的不足,本发明的目的是提供一种用于对不同樟树进行基因分型的snp引物,为29株樟树建立指纹图谱。本发明的另一目的是提供一种上述snp引物的应用。技术方案:为了实现上述发明目的,本发明采用的技术方案为:一种用于对不同樟树进行基因分型的snp引物,由8对引物组成,各引物的核苷酸序列如下表所示:引物名称上游引物(5’to3’)下游引物(5’to3’)snp1aagctgcaaacccattcgagtgacctgcttggcatacgcsnp2catggctctaatgggcaatctgcgggctctcgtagaaasnp3cgccaggcagggtaagccaacagacagggcactaaagsnp4gcctataagggataagagaatacattttacaaagagattccacatsnp5caaccctctaatcagtgatgagggtggatcttatcttgagatsnp6tgagttgaaactttatgaggccatctacttcagtcccaccttsnp7ctcaaaaggacaagagtgagatgtttgacttcagcagagactsnp8aagcttcttcttcgatatagactacgaaagaagaaagagaagaac应用所述的snp引物构建樟树的指纹图谱,获得29个樟树对应的指纹图谱,如下表所示:所述的应用snp引物构建樟树的指纹图谱,采用7对snp引物,名称分别为:snp1,snp2,snp3,snp4,snp5,snp6,snp7。所述snp引物在不同樟树基因组dna的snp位点基因分型的检测中的应用。所述的应用,包括以下步骤:1)待测樟树样品dna的提取;2)使用相同反应体系及程序对樟树样本基因组dna进行pcr扩增,得到待测樟树的pcr产物;3)对pcr扩增产物进行高分辨率溶解曲线分析确定snp位点的基因型;4)sanger测序再次验证snp基因型。步骤2)中,pcr反应体系20μl:30ng模板dna;10μl1xforget-me-nottmqpcrmastermix;0.1μmol/lsnp引物;剩余ddh2o补齐。步骤2)中,pcr反应程序:预变性95℃2min;95℃变性5s,45个循环;60℃复性30s。步骤3)中,hrm的反应程序:95℃10s,60℃1min,95℃15s,60℃15s。本发明通过实验证明:本发明的引物共8对,其中7对snp引物组合可以用于构建樟树的指纹图谱。本发明利用樟树数据开发的snp引物能够对不同樟树进行基因分型,并且本发明操作方法简单,通量高,成本低。此外,本发明开发的7对snp引物构建了不同樟树的指纹图谱,验证的8个snp位点还可以用来分子标记辅助育种,遗传多样性研究,基于snp的关联分析,以及用于种质资源鉴定与分类等。有益效果:与现有技术相比,本发明具有以下的技术优势:1)结果高度准确、可靠:利用本发明进行检测与鉴定,检测结果100%准确,检测结果提供了高度的可靠性。2)操作快速简便:进行不同个体间的样本检测,一般整个试验过程只需几小时即可完成,与传统的测序技术相比,省时省力,可以实现高通量。3)无需基于凝胶电泳进行,减少试剂和耗材的使用,更佳环保,同时节省人力和减少人工误差。4)检测结果灵敏度高:对待检测样品仅需提供模板dna30ng即可准确进行鉴定。5)效率高:相比于ssr指纹图谱,虽然snp单位点的理论信息量不如ssr丰富,但是snp在全基因组范围内密度远高于ssr,借助hrm分型可以迅速获取指纹信息,综合效率上高于ssr指纹。附图说明图1是以引物snp7为例,根据不同樟树个体表现出的差异溶解曲线峰,对其进行基因分型结果的溶解曲线,红色为cc,橘色为tt,蓝色为tc;图2是以引物snp2为例,pcr扩增产物的sanger测序结果图。具体实施方式下面结合具体实施例对本发明做进一步的说明。实施例11、初步筛选对樟树进行基因分型的snp引物1)转录组数据的获得从野外获得多份樟树样本,提取次生代谢产物,测定其生化类型,从其中选取芳樟型f-1、f-2以及龙脑型l-1、l-2樟树叶片样品,放于-80℃超低温冰箱保存备用。根据制造商的说明书使用rneasyplantminikit(qiagen,hilde,germany)进行樟树总rna提取。提取后,在1.0%的琼脂糖凝胶上检测rna的降解和污染。使用分光光度计(implen,westlakevillage,ca,usa)检查rna纯度。使用2.0flurometer(lifetechnologies,carlsbad,ca,usa)中的rnaassaykit测定rna的浓度。使用anagilentbioanalyzer2100system(agilenttechnologies,santaclara,ca,usa)中的rnanano6000assaykit评估rna的完整性。每个样品的总量为3μgrna用作rna样品制备的输入材料。按照制造商的说明书,使用ultratmrnalibraryprepkit(neb,usa)生成测序文库,并将索引代码添加到每个样品的属性序列中。简而言之,使用poly-toligo附着磁珠从总rna纯化mrna。在nebnextfirststrandsynthesisreactionbuffer(5x)中,在高温下使用二价阳离子打断成短片段。以mrna为模板,使用六碱基随机引物和m-mulv逆转录酶(rnaseh-)合成第一链cdna。随后使用dna聚合酶i和rna酶h进行第二链cdna合成。通过外切核酸酶/聚合酶活性将剩余突出部分转化为平端。在dna片段的3'末端腺苷酸化后,连接具有发夹环结构的nebnext适配子以进行杂交。为了选择长度优先为150-200bp的cdna片段,文库片段用ampurexp系统(beckmancoulter,beverly,usa)纯化。然后将3μluserenzyme(neb,usa)与大小选择的接头连接的cdna在37℃下使用15min,然后在95℃下pcr5min。进而使用phusion高保真dna聚合酶,通用pcr引物和index(x)引物进行pcr。最后,将pcr产物纯化(ampurexp系统),并在agilentbioanalyzer2100系统上评估文库质量。根据制造商的说明书,使用truseqpeclusterkitv3-cbot-hs(illumia)在cbot群集生成系统上进行索引编码样本的聚类。集群生成后,在illuminahiseqtm2500高通量平台上对其准备进行测序,并生成配对末端读取。fastq格式的原始数据(rawreads)首先通过内部perl脚本进行处理。在此步骤中,通过删除包含适配器的读取,包含poly-n的读取以及从原始数据读取的低质量来获取干净的数据(cleanreads)。同时,计算了cleandata的q20,q30,gc含量和序列重复水平。所有的下游分析都是以高质量的cleandata为基础。然后,将所有库/样本中的左文件(read1files)合并到一个大的left.fq文件中,将正确的文件(read2files)合并到一个大的right.fq文件中。基于left.fq和right.fq使用trinity完成转录组组装,默认情况下min_kmer_cov设置为2,其他所有参数设置为默认值。选择具有最长长度的每个基因的转录本作为unigenes。1.snp的识别本实施例通过使用picard-toolsv1.41和samtoolsv0.1.18来排序,删除重复读取,对齐合并获得每个样本的bam结果。采用gatk2v3.2软件用于执行snpcalling(mckennaetal,2010)。原始的vcf文件使用gatk标准方法过滤(参数为clusterwindowsize:10;mq0>=4and(mq0/(1.0*dp))>0.1;qual<10;qual<30.0orqd<5.0orhrun>5),最后仅保留距离大于5的snp。2)snp引物的设计根据转录组unigene序列,挑选60个snp位点,利用oligo6.0软件设计60对snp引物。设计的方法如下:首先确定snp位点所在序列的位置,然后在snp位点的上下游分别设计正向引物和反向引物,引物设计的参数为引物长度18-23bp;tm59℃-61℃,尽可能保证上下游引物的tm值一致,一般不超过2℃;gc含量在40-60%(45-55%最佳);pcr产物大小为150-400bp;其它还应注意引物3'端不应出现超过3个的连续g或c;引物不能形成发夹结构,即不能有4bp以上的回文序列;引物自身、引物之间尽量不能有连续4个以上的碱基互补,尤其避免3'端的互补。3)ctab法提取樟树基因组dna:打开水浴锅,调节至65℃预热备用。将50-100mg的樟树叶片在液氮中充分研磨成粉末,并转移只1.5ml的离心管中。向离心管中加入4mlctab,80μl120%巯基乙醇。放入65℃水浴锅中预热30min后取出,置于冰水中冷却至室温。加入4ml氯仿(异戊二醇:氯仿=1:24),12000rpm,离心4min。取上清至一干净离心管中,加入3ml氯仿,抽提第二次,12000rpm,离心4min。将上清分装在四个离心管中(1个2ml离心管装备用液、3个1.5ml离心管),每个1.5ml离心管分装500μl。向每个离心管中加入2倍体积(1000μl)冰无水乙醇,再加50μl醋酸钠,放置在-20℃冰箱,2h后取出,若无絮状物,12000rpm离心5min。准备一个新的1.5ml离心管,加入1000μl70%乙醇。将三个离心管中的絮状物全部用枪头挑出,加入新的离心管中。涡旋,12000rpm离心2min。再取一新的离心管,第二次用70%乙醇漂洗、涡旋、离心。小心的吸出离心管中的酒精,注意不要吸到絮状物。加热干燥,使酒精全部蒸发。向离心管中加入200μlddh2o,溶解1h,置于-20℃冰箱保存备用。4)snp引物的有效性验证以樟树基因组dna为模板,采用待检测snp引物进行pcr扩增,获得pcr扩增产物;反应结束后,取3μl的pcr产物加入4μlloadingbuffer,再加3μlddh2o,利用8%聚丙烯酰胺凝胶进行电泳,然后采用银染方法对pcr扩增产物进行检测。能检测到目的条带且无其它杂带和引物二聚体的,该待检测引物即为有效引物。10μl的la-taq酶pcr反应体系如下:5μl的premixtaq(lataqversion2.0plusdye);0.2μmol/lsnp引物;40ngdna模版;剩余ddh2o补齐。pcr的反应程序为:预变性94℃3min;35循环的94℃30s,最佳tm55℃30s,72℃30s;延伸72℃1min,最终4℃保存。结果表明:挑选的60对snp引物中,有45对引物能够扩增出条带,但其中的35对引物扩增出的条带不单一,可能存在多个snp位点,暂不考虑进行hrm分析,另外10对引物(见下表)扩增条带单一且产物大小与预期一致,暂为有效snp引物,用于hrm分析。1对引物对应1个目标snp位点,1对引物的扩增产物为含有对应snp位点的dna片段。2、再次筛选对樟树进行基因分型的snp引物1)pcr扩增及高分辨率溶解曲线(hrm)分析进行基因分型以33个樟树个体(其中4个樟树个体为转录组测序的樟树,芳樟型f-1、f-2和龙脑型l-1、l-2;其余29棵樟树来源见表1)的基因组dna为模板,分别采用上述10对snp有效性引物进行pcr扩增,得到每个个体的10个引物对应的pcr扩增产物,每个引物对的pcr扩增产物为含有某个snp位点的dna片段。pcr反应体系20μl:30ng模板dna;1xforget-me-nottmqpcrmastermix10μl;0.1μmol/lsnp引物;剩余ddh2o补齐。采用viia7中的highresolutionmelt进行pcr及基因分型。第一步,10μl的pcr反应程序:预变性95℃2min;45个循环的95℃变性5s,60℃复性30s;第二步,hrm的反应程序:95℃10s,60℃1min,95℃15s,60℃15s。结果表明:有8对引物能够进行基因分型,其余2对引物因有杂峰(snp9,snp10),所以被淘汰。采用appliedhrmsoftwarev2.0进行溶解曲线分析。8对引物对应的snp位点得到hrm验证,其中,高分辨率溶解曲线部分结果如下:图1为以snp引物7为例,根据不同樟树个体表现出的差异溶解曲线峰,对其进行基因分型结果的溶解曲线,红色为cc,橘色为tt,蓝色为tc。8对snp引物对樟树个体的基因分型结果如表2所示:表1樟树材料来源樟树样品号采集地樟树样品号采集地y-1江西崇义y-16江西崇义y-2江西安福y-17湖南郴州y-3江西龙南y-18四川宜宾y-4江西分宜y-19四川达州y-5江西资溪y-20四川成都y-6江西广丰y-21四川广元y-7江西广昌y-22湖北黄冈y-8江西瑞昌y-23湖北襄阳y-9江西瑞金y-24江苏南通y-10江西信丰y-25江苏连云港y-11江西乐安y-26江苏徐州y-12江西赣县y-27江苏盐城y-13江西丰城y-29福建龙岩y-14湖南长沙y-29福建厦门y-15湖南长沙表2樟树样本分型结果其中,7对snp引物组合(snp1-7)构建樟树指纹代码,获得29个香樟对应的指纹图谱,如表3所示。表329个香樟对应的指纹图谱样品号snp指纹代码样品号snp指纹代码y-15221543y-163515254y-23515553y-171511245y-33551545y-185211243y-45511553y-193521435y-53255543y-201521253y-63521243y-215213253y-75555253y-221115253y-83551543y-235155253y-93525243y-245511253y-105525254y-255555543y-111221233y-265255253y-125211233y-271523243y-133215243y-281551545y-141215233y-291551535y-151515243注:纯合型aa,gg,tt,cc对应1,2,3,4,突变型对应5。实施例2高分辨率溶解曲线分析结果的验证通过pcr产物测序对高分辨率溶解曲线分析得到的基因分型的结果进行验证。具体步骤如下:挑选snp的不同分型的pcr产物20μl,进行sanger测序,根据测序峰图和等位基因出现的频率确定基因型,并与hrm基因分型结果进行比较,以验证引物基因分型的准确性。经过sanger测序验证本发明的引物通过hmr分析对银杏进行基因分型的结果是可靠的。测序结果如下:通过测序获得基因峰图,以snp2引物为例(图2的a,b),其扩增片段的snp位点为g/a突变,图2a基因型为a/t,图2b基因型为a/a,均符合hrm基因分型结果,认为snp为阳性snp位点且hrm基因分型结果准确。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1