N‑烷基/烯基磺酰亚胺衍生物、其制备方法及应用与流程
本发明属于有机合成化学技术领域,特别涉及n-烷基/烯基磺酰亚胺衍生物的制备方法和应用。
背景技术:
磺酰胺类化合物在医药和农药上具有广泛的生物活性,是近年来有机合成研究的热点之一。因此,把磺酰胺引入到不同的化合物结构中,通过结构修饰可以产生一系列具有生物活性的化合物。特别是,通过羧酸类化合物的施密特反应形成新的c-n键(k.f.schmidt,ber.,1924,57b,704),是构建胺、酰胺以及磺酰胺类化合物的最有效途径之一。然而,公开的传统的方法存在一些缺陷,例如:使用具有爆炸性的叠氮类化合物作为氮源、使用腐蚀性较强的磺酸和高毒的有机试剂。因此,开发一种清洁、简便的合成磺酰胺类化合物的方法具有重要意义。
近年来,许多科学家利用脱羧c-c、c-p以及c-x偶联反应构建许多具有生物、药物活性的功能性化合物(science,2006,313,662;chem.soc.rev.,2011,40,5030;j.am.chem.soc.,2012,134,10401)。众所周知,通过脱羧c-n偶联反应是在反应物分子中引入氨基最有效的条途径之一。但是,过渡金属催化的脱羧c-n偶联反应报道较少。其中,lei课题组首次发现了在光催化体系实现了α-羰基酸与芳基酰胺的自由机型的脱羧c(sp2)–n偶联反应。紧接着,mainolfi和jia课题组进一步发展了脱羧c(sp2)–n偶联反应。随后,jiao和bolm研究小组开发了铜催化的脱羧c(sp)–n偶联反应。因此,开发同时实现脱羧c(sp2)–n偶联反应和脱羧c(sp3)–n偶联反应是一个具有挑战的课题。到目前为止,同时实现铜催化的羧酸类化合物和n-氟代二苯磺酰亚胺合成磺酰亚胺类化合物的脱羧c(sp2)–n偶联反应和脱羧c(sp3)–n偶联反应还未见文献报道。
技术实现要素:
针对现有技术中存在的问题,本发明通过以1,10-邻菲罗啉作为配体,在催化剂作用下由羧酸类化合物和n-氟代二苯磺酰亚胺经过分子间脱羧偶联反应,制备一系列的n-烷基/烯基磺酰亚胺类化合物。
本发明提供的n-烷基/烯基磺酰亚胺衍生物,具有通式(ⅰ)所示的结构:
其中:r选自碳原子数为1~6的烷基或烯基,这些烷基和烯基分别独立地被苯基、取代的苯基、含s原子的五元杂环、萘基所取代。
优选地,所述n-烷基/烯基磺酰亚胺衍生物包括如下化合物:
本发明提供的n-烷基/烯基磺酰亚胺衍生物的制备方法,具体过程如下:
以通式(ⅱ)所示的羧酸类化合物和n-氟代二苯黄酰亚胺为原料,1,10-邻菲罗啉作为配体,在催化剂的作用下,在非水溶剂中进行分子间脱羧偶联反应得到通式(ⅰ)所示的n-烷基/烯基磺酰亚胺衍生物,其反应方程式如下所示:
其中,通式(ⅱ)中r的定义同上。
优选地,所述催化剂用量为0.1~150mol%。
更优选地,所述催化剂用量为10~20mol%。
优选地,所述催化剂选自四氯化钛、三氯化铁、溴化铜、碘化亚铜、醋酸铜、三氟化硼乙醚、三氟甲磺酸、硫酸中的至少一种。
优选地,所述非水溶剂选自苯、二甲苯、甲苯、四氢呋喃、1,4-二氧六环、乙腈、二甲亚砜、n,n-二甲基甲酰胺、乙醇中的至少一种。
优选地,反应温度为30~150℃。
本发明提供的n-烷基/烯基磺酰亚胺衍生物作为杀虫剂的应用。
与现有技术相比,本发明具有如下有益效果:
(1)本发明实施例以羧酸类化合物和n-氟代二苯黄酰亚胺为原料,1,10-邻菲罗啉作为配体,在铜催化剂的作用下,在非水溶剂中进行分子间脱羧偶联反应得到n-烷基/烯基磺酰亚胺衍生物,同时实现了脱羧c(sp2)–n偶联反应和脱羧c(sp3)–n偶联反应。
(2)本发明实施例提供的方法能够高效地单一选择性的制备高纯度的n-烷基/烯基磺酰亚胺衍生物,经生物活性测试结果表明,本发明实施例合成的n-烷基/烯基磺酰亚胺衍生物具有很好的杀虫效果,能够广泛应用与动物学、植物学以及医学等诸多领域。
附图说明
图1为本发明实施例4提供的化合物的1h-nmr的核磁共振谱图;
图2为本发明实施例4提供的化合物的13c-nmr的核磁共振谱图;
图3为本发明实施例5提供的化合物的1h-nmr的核磁共振谱图;
图4为本发明实施例5提供的化合物的13c-nmr的核磁共振谱图。
具体实施方式
为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例对本发明作进一步说明,但所举实施例不作为对本发明的限定。
需要说明的是,下面实施例中未注明具体条件的实验方法,均按照本领域的常规方法和条件进行,所用试剂如无特殊说明,均为常规试剂。
实施例1
化合物(1)的制备,具体反应方程式如下所示:
具体制备过程为:向带有磁力搅拌装置的10ml带支口圆底烧瓶中加入乙腈(2ml)作为溶剂、4-甲基苯乙酸(0.46g,1mmol)、n-氟代二苯磺酰亚胺(0.097g,1.2mmol)、碘化亚铜(0.026ml,0.3mmol)和1,10-邻菲罗啉(0.026ml,0.3mmol),搅拌均匀后,将其放入80℃油浴中继续搅拌。tlc检测底物消失,反应结束。将反应液倾入饱和氯化钠水溶液(5ml)中,用二氯甲烷(3×6ml)萃取,合并有机相,无水硫酸镁干燥、抽虑,然后减压蒸馏除去有机溶剂,得到液体混合物,最后经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=10:1)得到淡黄色固体0.41g,经过nmr、ms证实为化合物(1),以4-甲基苯乙酸为基础,计算其收率为90%。
化合物(1)的谱图解析数据具体如下:
1hnmr(500mhz,cdcl3)δ7.78(d,j=7.5hz,4h),7.56(t,j=7.5hz,2h),7.42(t,j=8.0hz,4h),7.26-7.23(m,2h),7.03(d,j=7.5hz,2h),4.89(s,2h),2.32(s,3h);
13cnmr(125mhz,cdcl3)δ140.0,137.8,133.4,131.4,129.1,128.9,128.7,128.0,52.2,21.0;
hrms(esi)m/zcalculatedforc20h19nnao4s2[m+na]+:424.0653,found424.0658。
实施例2
化合物(2)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“4-氟苯乙酸”代替“4-甲基苯乙酸”,且反应温度为30℃,反应时间为6h。经过nmr、ms证实为化合物(2),以4-氟苯乙酸为基础,计算其收率为89%。
化合物(2)的谱图解析数据具体如下:
1hnmr(500mhz,cdcl3)δ7.80(d,j=7.5hz,4h),7.60(t,j=7.5hz,2h),7.46(t,j=8.0hz,4h),7.37(dd,j=5.5,8.5hz,2h),6.93(t,j=9.0hz,2h),4.89(s,2h);
13cnmr(125mhz,cdcl3)δ163.5(d,j=245.7hz),139.8,133.6,131.1(d,j=8.1hz),130.3(d,j=2.5hz),128.8,128.0,115.3(d,j=21.3hz),51.6;
hrms(esi)m/zcalculatedforc19h16fnnao4s2[m+na]+:428.0402,found428.0407。
实施例3
化合物(3)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“2-噻吩乙酸”代替“4-甲基苯乙酸”,溶剂选择1,2-二氯乙烷,且反应温度为130℃,反应时间为8h。经过nmr、ms证实为化合物(3),以2-噻吩乙酸为基础,计算其收率为91%。
化合物(3)的谱图解析数据具体如下:
1hnmr(300mhz,cdcl3)δ7.88-7.81(m,4h),7.63-7.56(m,2h),7.50-7.42(m,4h),7.25(dd,j=1.2,5.1hz,1h),7.12(d,j=2.7hz,1h),6.92(dd,j=3.6,5.1hz,1h),5.09(s,2h);
13cnmr(75mhz,cdcl3)δ139.8,137.2,133.7,129.4,128.8,127.9,127.2,126.4,47.1;
hrms(esi)m/zcalculatedforc17h15nnao4s3[m+na]+:416.0061,found416.0061。
实施例4
化合物(4)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“2-萘乙酸”代替“4-甲基苯乙酸”,溶剂为乙醇,且反应温度为80℃,反应时间为10h。经过nmr、ms证实为化合物(4),以2-萘乙酸为基础,计算其收率为73%。
化合物(4)的谱图解析数据具体如下:
1hnmr(300mhz,cdcl3)δ8.09(d,j=8.4hz,1h),7.81-7.72(m,5h),7.69(d,j=8.4hz,1h),7.53-7.39(m,4h),7.38-7.30(m,5h),7.25-7.17(m,1h),5.52(s,2h);
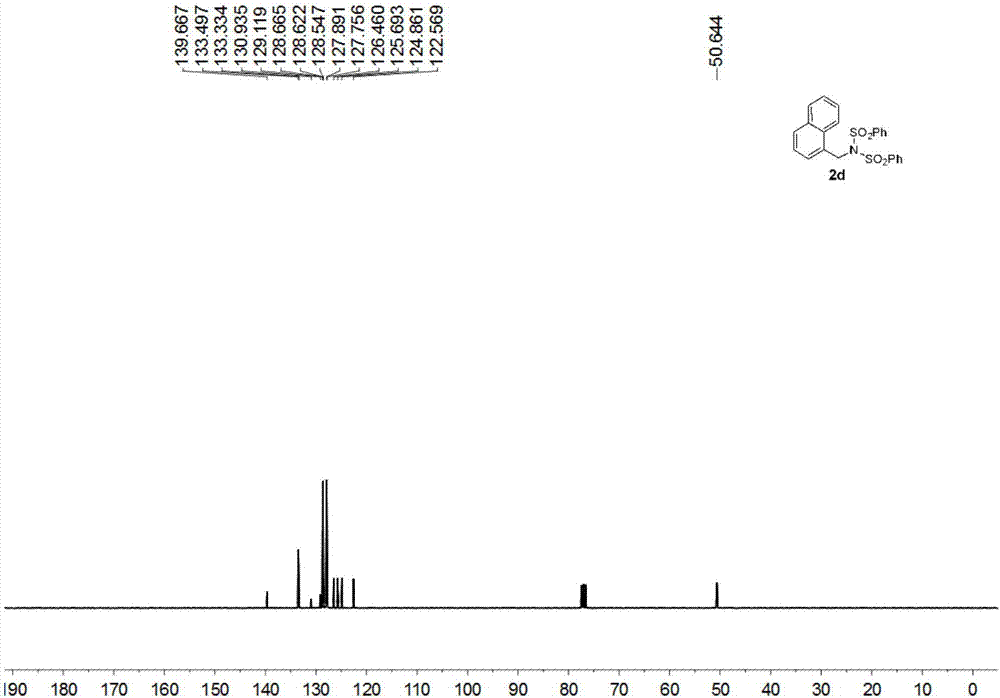
13cnmr(75mhz,cdcl3)δ139.6,133.4,133.3,130.9,129.1,128.66,128.62,128.5,127.8,127.7,126.4,125.6,124.8,122.5,50.6;
hrms(esi)m/zcalculatedforc23h19nnao4s2[m+na]+:460.0653,found460.0654。
化合物(4)的1h-nmr的核磁共振谱图和13c-nmr的核磁共振谱图见图1和图2;
实施例5
化合物(5)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“4-甲氧基肉桂酸”代替“4-甲基苯乙酸”,溶剂为n,n-二甲基甲酰胺,且反应温度为100℃,反应时间为12h。经过nmr、ms证实为化合物(5),以4-甲氧基肉桂酸为基础,计算其收率为98%。
化合物(5)的谱图解析数据具体如下:
1hnmr(400mhz,cdcl3)δ8.00(d,j=8.0hz,4h),7.67(t,j=7.2hz,2h),7.56(t,j=8.0hz,4h),7.30(d,j=8.4hz,2h),6.87(d,j=8.4hz,2h),6.61(d,j=13.6hz,1h),6.38(d,j=13.6hz,1h),3.81(s,3h);
13cnmr(100mhz,cdcl3)δ160.5,139.5,139.2,133.8,129.0,128.6,128.1,126.2,117.0,114.1,55.3;
hrms(esi)m/zcalculatedforc21h20no5s2[m+na]+:430.0783,found430.0785。
化合物(5)的1h-nmr的核磁共振谱图和13c-nmr的核磁共振谱图见图1和图2;
实施例6
化合物(6)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“4-氟肉桂酸”代替“4-甲基苯乙酸”,溶剂为甲苯,催化剂为氯化亚铜,且反应温度为100℃,反应时间为12h。经过nmr、ms证实为化合物(6),以4-氟肉桂酸为基础,计算其收率为88%。
化合物(6)的谱图解析数据具体如下:
1hnmr(500mhz,cdcl3)δ8.01-7.97(m,4h),7.69(t,j=7.5hz,2h),7.57(t,j=8.0hz,4h),7.36-7.31(m,2h),7.07-7.01(m,2h),6.66(d,j=13.5hz,1h),6.46(d,j=13.5hz,1h);
13cnmr(125mhz,cdcl3)δ164.3(d,j=248.7hz),139.4,137.8,134.0,129.9(d,j=3.3hz),129.1,129.0(d,j=12.5hz),128.1,119.1(d,j=1.6hz),115.9(d,j=21.7hz);
hrms(esi)m/zcalculatedforc20h16fnnao4s2[m+na]+:440.0402,found440.0404。
实施例7
化合物(7)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“2,4-二氯肉桂酸”代替“4-甲基苯乙酸”,溶剂为二甲亚砜,且反应温度为150℃,反应时间为16h。经过nmr、ms证实为化合物(7),以2,4-二氯肉桂酸为基础,计算其收率为82%。
化合物(7)的谱图解析数据具体如下:
1hnmr(400mhz,cdcl3)δ8.02(d,j=7.6hz,4h),7.71(t,j=7.6hz,2h),7.60(t,j=8.0hz,4h),7.42-7.38(m,2h),7.24(dd,j=1.2,8.4hz,1h),7.00(d,j=13.6hz,1h),6.55(d,j=13.6hz,1h);
13cnmr(100mhz,cdcl3)δ139.3,135.4,134.4,134.1,133.2,130.8,129.7,129.1,128.2,128.1,127.4,122.4;
hrms(esi)m/zcalculatedforc20h15cl2nnao4s2[m+na]+:489.9717,found489.9718。
实施例8
化合物(8)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“2-噻吩烯丙酸”代替“4-甲基苯乙酸”,溶剂为二甲亚砜,催化剂为无水醋酸铜,且反应温度为150℃,反应时间为18h。经过nmr、ms证实为化合物(8),以2-噻吩烯丙酸为基础,计算其收率为93%。
化合物(8)的谱图解析数据具体如下:
1hnmr(400mhz,cdcl3)δ7.93(d,j=7.6hz,4h),7.62(t,j=7.6hz,2h),7.51(t,j=7.6hz,4h),7.23(d,j=4.8hz,1h),7.00(d,j=3.2hz,1h),6.96-6.93(m,1h),6.78-6.72(m,1h),6.32(d,j=13.6hz,1h);
13cnmr(100mhz,cdcl3)δ139.4,137.3,134.0,132.3,129.1,128.8,128.2,127.7,126.8,117.9;
hrms(esi)m/zcalculatedforc18h15nnao4s3[m+na]+:428.0061,found428.0064。
实施例9
化合物(9)的制备,具体反应方程式如下所示:
具体制备过程和实施例1相同,不同之处仅在于,用“1-萘烯丙酸”代替“4-甲基苯乙酸”,溶剂为二甲亚砜,催化剂为三氟甲磺酸铜,且反应温度为150℃,反应时间为20h。经过nmr、ms证实为化合物(9),以1-萘烯丙酸为基础,计算其收率为87%。
化合物(9)的谱图解析数据具体如下:
1hnmr(400mhz,cdcl3)δ8.06(d,j=8.0hz,4h),7.89-7.82(m,3h),7.70(t,j=7.6hz,2h),7.59(t,j=7.6hz,5h),7.54-7.50(m,2h),7.48-7.42(m,2h),6.53(d,j=13.2hz,1h);
13cnmr(100mhz,cdcl3)δ139.5,137.3,134.0,133.4,131.2,131.1,129.6,129.1,128.5,128.2,126.7,126.2,125.4,124.8,123.7,121.6;
hrms(esi)m/zcalculatedforc24h19nnao4s2[m+na]+:472.0653,found472.0656。
上述实施例1-实施例9制备得到的化合物具有很好的杀虫效果,可作为杀虫剂来使用,为了证明其效果,下面以部分化合物为例,通过东方粘虫试验,对这些化合物的杀虫效果进行进一步验证。
具体的,下述表1中化合物分别配置不同浓度的水溶液,在室温条件下,当相对湿度为80%,对东方粘虫进行生物活性实验,这些化合物对东方粘虫的抑制率如下表1所示:
表1不同浓度的各化合物对东方粘虫的抑制率
由表1可以看出,随着浓度的增大,对东方粘虫的抑制率逐渐增大,其中化合物(1)浓度为100mg/l时,杀东方粘虫的生物活性可达67%,其他化合物均对东方粘虫有不同程度的抑制情况,说明本发明实施例1-实施例9提供的化合物是一种潜在的杀虫剂,可作为杀虫估计来使用,有效预防和治疗农作物的病虫害。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,其保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内,本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!