巴瑞克替尼磷酸盐I晶型及其制备方法与流程
本申请是2016年2月1日提交的申请号为201610070654.0的中国发明专利申请《巴瑞克替尼磷酸盐的a晶型、h晶型和i晶型及其制备方法》的分案申请。本发明涉及作为jak抑制剂的巴瑞克替尼衍生物的多晶型,具体地,涉及巴瑞克替尼磷酸盐的a晶型、h晶型和i晶型及其制备方法。
背景技术:
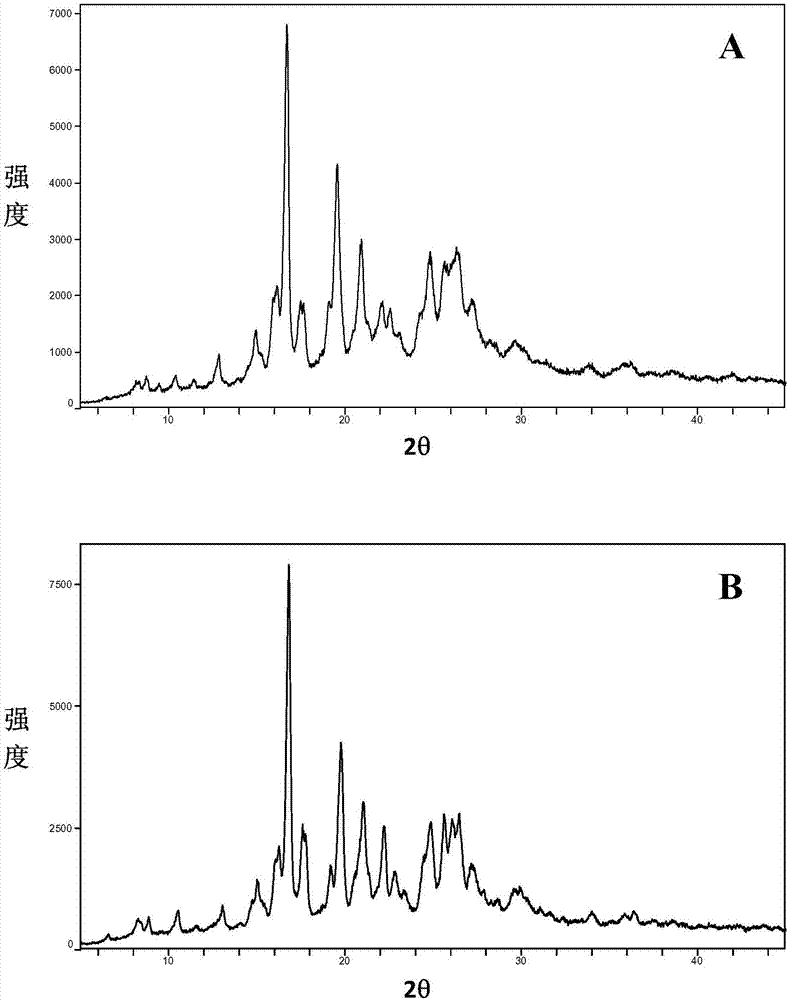
:jak即两面神激酶(januskinase),是一种非受体型酪氨酸蛋白激酶,也是一类非跨膜型的酪氨酸激酶。这是因为jak既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定sh2结构域的信号分子。jak蛋白家族共包括4个成员:jak1、jak2、jak3以及tyk2,它们在结构上有7个jak同源结构域(jakhomologydomain,jh),其中jh1结构域为激酶区、jh2结构域是“假”激酶区、jh6和jh7是受体结合区域。tyk2是免疫炎性疾病的潜在靶点,已经通过人遗传学和小鼠剔除研究确证(levyd.和loomisc.,newenglandjournalofmedicine357(2007年)1655-1658页)。jak1是免疫炎性疾病领域的新靶点。将jak1与其它jak杂二聚化,能转导细胞因子驱动的促炎信号传导。因此,预期抑制jak1和或其它jak对于一系列炎性病症和其它由jak介导的信号转导驱动的疾病是有治疗益处的。本发明中的巴瑞克替尼(baricitinib)磷酸盐是化学名为{1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]氮杂环丁烷-3-基}乙腈磷酸盐,如式(i)所示的化合物,其是一种jak类抑制剂化合物,可用于治疗jak参与的自身免疫疾病、炎性疾病或癌症疾病。在中国发明专利cn102026999(wo2009/114512)中,公开了该化合物及其衍生物。按照此专利重复得到固体,熔点为188℃,和专利公开类似,本发明中将其称为“晶型x巴瑞克替尼磷酸盐”。众所周知,同一种药物,晶型不同,其生物利用度也可能会存在差别,另外其稳定性、流动性、可压缩性也可能会不同,这些理化性质对药物的应用产生一定的影响,从而影响药物的疗效。因此,需要具有优越的生理化学特性的巴瑞克替尼衍生物的晶型,其可有利地在药物加工和药物组合物中使用。本发明研制的巴瑞克替尼磷酸盐的新晶型未见报道。技术实现要素:本发明所要解决的问题是现有巴瑞克替尼磷酸盐溶解度有待提高而影响生物利用度的问题,同时希望能够寻求巴瑞克替尼磷酸盐的新晶型,为固体药物的疗效研究提供更多的定性定量信息。为了解决上述技术问题,本发明的第一个方面提供了一种巴瑞克替尼磷酸盐的新的晶型,更具体地,为{1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]氮杂环丁烷-3-基}乙腈磷酸盐的新的晶型(以下称“巴瑞克替尼磷酸盐a晶型”),其xrpd图谱在2θ=8.38、8.88、10.48、13.00、15.06、16.22、16.82、17.76、19.18、19.70、21.04、22.22、22.68、24.40、24.94、26.48、27.40处有衍射峰,其中2θ值误差范围为±0.2。本发明所述的巴瑞克替尼磷酸盐a晶型,具有与说明书附图图1基本上相同的xrpd图谱。本发明还提供了制备巴瑞克替尼磷酸盐a晶型的一种方法,包括以下步骤:(1)在巴瑞克替尼磷酸盐中加入用于溶解的有机溶剂而得到所述巴瑞克替尼磷酸盐的溶解物;(2)将用于析出的有机溶剂加入所述巴瑞克替尼磷酸盐的溶解物中,使所述巴瑞克替尼磷酸盐析出,得到所述巴瑞克替尼磷酸盐的析出物;(3)将所述巴瑞克替尼磷酸盐的析出物干燥,得到巴瑞克替尼磷酸盐a晶型。在一些实施例中,制备所述杂环丁烷磷酸盐a晶型的方法还包括对所述巴瑞克替尼磷酸盐的析出物进行过滤的步骤。在一些实施例中,所述用于溶解的有机溶剂为酰胺类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述用于溶解的有机溶剂为n,n二甲基甲酰胺。在一些实施例中,所述用于析出的有机溶剂为醚类或酯类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述醚类有机溶剂为乙醚、异丙醚或甲基叔丁基醚;所述酯类有机溶剂为乙酸乙酯或乙酸异丙酯。在一些实施例中,在所述巴瑞克替尼磷酸盐析出之后,继续加入所述用于析出的有机溶剂。在一些实施例中,所述用于溶解的有机溶剂与所述用于析出的有机溶剂的体积比例为1:2至1:15。在一些实施例中,将所述析出物真空干燥而得到所述巴瑞克替尼磷酸盐a晶型。在一些实施例中,所述巴瑞克替尼磷酸盐于室温下溶解或析出。此外,本发明的第二个方面提供了一种巴瑞克替尼磷酸盐的新的晶型,更具体地,为{1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]氮杂环丁烷-3-基}乙腈磷酸盐的新的晶型(以下称“巴瑞克替尼磷酸盐h晶型”),其xrpd图谱在2θ=7.94、12.401、13.539、14.278、15.957、16.779、17.52、19.403、20.282、22.259、24.617、26.04、27.144处有衍射峰,其中2θ值误差范围为±0.2。本发明所述的巴瑞克替尼磷酸盐h晶型,具有与说明书附图图6基本上相同的xrpd图谱。本发明还提供了制备巴瑞克替尼磷酸盐h晶型的一种方法,包括以下步骤:(1)在巴瑞克替尼磷酸盐中加入用于溶解的有机溶剂而得到所述巴瑞克替尼磷酸盐的溶解物;(2)将用于析出的有机溶剂加入所述巴瑞克替尼磷酸盐的溶解物中,使所述巴瑞克替尼磷酸盐析出,得到所述巴瑞克替尼磷酸盐的析出物;其中,所述用于析出的有机溶剂温度低于室温;(3)将所述巴瑞克替尼磷酸盐的析出物干燥,得到巴瑞克替尼磷酸盐h晶型。在一些实施例中,制备所述杂环丁烷磷酸盐h晶型的方法还包括对所述巴瑞克替尼磷酸盐的析出物进行过滤的步骤。在一些实施例中,所述用于溶解的有机溶剂为酰胺类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述用于溶解的有机溶剂为n,n二甲基甲酰胺。在一些实施例中,所述用于析出的有机溶剂为酯类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述酯类有机溶剂为乙酸乙酯。在一些实施例中,所述用于析出的有机溶剂的温度为-1至3℃,并且在将所述用于析出的有机溶剂加入至所述溶解物中之后,将所述析出物在-1至3℃下保持。在一些实施例中,将所述析出物真空干燥而得到所述巴瑞克替尼磷酸盐h晶型。本发明还提供了制备巴瑞克替尼磷酸盐h晶型的另一种方法,包括以下步骤:(1)在巴瑞克替尼中加入用于溶解的有机溶剂而得到所述巴瑞克替尼的溶解物;其中,所述用于溶解的有机溶剂的温度高于室温;(2)将含磷酸的有机溶剂加入所述巴瑞克替尼的溶解物中,获得巴瑞克替尼磷酸盐的溶解物;(3)将所述巴瑞克替尼磷酸盐的溶解物的温度迅速降至室温,使所述巴瑞克替尼磷酸盐析出,得到所述巴瑞克替尼磷酸盐的析出物;(4)将所述巴瑞克替尼磷酸盐的析出物干燥,得到巴瑞克替尼磷酸盐h晶型。在一些实施例中,制备所述杂环丁烷磷酸盐h晶型的方法还包括对所述巴瑞克替尼磷酸盐的析出物进行过滤的步骤。在一些实施例中,所述用于溶解的有机溶剂为腈类或醇类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述腈类有机溶剂为乙腈;所述醇类有机溶剂为乙醇。在一些优选的实施例中,所述用于溶解的有机溶剂为乙腈和乙醇的混合溶剂。在一些优选的实施例中,所述乙腈与所述乙醇的体积比例为3:1至5:1。在一些更优选的实施例中,所述乙腈与所述乙醇的体积比例为25:8。在一些实施例中,所述用于溶解的有机溶剂的温度为65℃至85℃。在一些优选的实施例中,所述用于溶解的有机溶剂的温度为80℃。在一些实施例中,所述含磷酸的有机溶剂为含磷酸的乙醇溶液。在一些实施例中,所述步骤(3)中的降温方法为将所述巴瑞克替尼磷酸盐的溶解物通入20-30℃的降温介质条件来降温。在一些实施例中,将所述析出物真空干燥而得到所述巴瑞克替尼磷酸盐h晶型。此外,本发明的第三个方面提供了一种巴瑞克替尼磷酸盐的新的晶型,更具体地,为{1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]氮杂环丁烷-3-基}乙腈磷酸盐的新的晶型(以下称“巴瑞克替尼磷酸盐i晶型”),其xrpd图谱在2θ=8.818、10.442、12.901、14.901、16.076、16.88、17.54、18.677、19.12、19.44、20.596、21.699、22.422、23.181、24.539、25.779、27、28.461处有衍射峰,其中2θ值误差范围为±0.2。本发明所述的巴瑞克替尼磷酸盐i晶型,具有与说明书附图图11基本上相同的xrpd图谱。本发明还提供了制备巴瑞克替尼磷酸盐i晶型的一种方法,包括以下步骤:(1)在巴瑞克替尼中加入用于溶解的有机溶剂而得到所述巴瑞克替尼的溶解物;其中,所述用于溶解的有机溶剂的温度高于室温;(2)将含磷酸的有机溶剂加入所述巴瑞克替尼的溶解物中,获得巴瑞克替尼磷酸盐的溶解物;(3)将所述巴瑞克替尼磷酸盐的溶解物的温度缓慢降至室温,使所述巴瑞克替尼磷酸盐析出,得到所述巴瑞克替尼磷酸盐的析出物;(4)将所述巴瑞克替尼磷酸盐的析出物干燥,得到巴瑞克替尼磷酸盐i晶型。在一些实施例中,制备所述杂环丁烷磷酸盐i晶型的方法还包括对所述巴瑞克替尼磷酸盐的析出物进行过滤的步骤。在一些实施例中,所述用于溶解的有机溶剂为腈类或醇类中任意一种溶剂或者两种以上溶剂以任意比例的混合溶剂。在一些优选的实施例中,所述腈类有机溶剂为乙腈;所述醇类有机溶剂为乙醇。在一些优选的实施例中,所述用于溶解的有机溶剂为乙腈和乙醇的混合溶剂。在一些优选的实施例中,所述乙腈与所述乙醇的体积比例为3:1至5:1。在一些更优选的实施例中,所述乙腈与所述乙醇的体积比例为25:8。在一些实施例中,所述用于溶解的有机溶剂的温度为65℃至85℃。在一些优选的实施例中,所述用于溶解的有机溶剂的温度为80℃。在一些实施例中,所述含磷酸的有机溶剂为含磷酸的乙醇溶液。在一些实施例中,所述步骤(3)中的降温方法为在80-100分钟内将所述巴瑞克替尼磷酸盐的溶解物的温度从所述高于室温的温度降低至室温。在一些实施例中,将所述析出物真空干燥而得到所述巴瑞克替尼磷酸盐i晶型。本领域普通技术人员可以根据其知识和经验,对本发明方法所用试剂的用量进行调整,包括按比例放大或缩小原料用量和调整溶剂用量,并且可以改变本发明方法的温度。这些调整的方案也包含在本发明的方法中。本发明所述的巴瑞克替尼磷酸盐a晶型、h晶型和i晶型,其稳定性与现有的晶型x巴瑞克替尼磷酸盐相当,且具有高于晶型x巴瑞克替尼磷酸盐的溶解度,可提升其生物利用度,有利于其药物加工和在药物组合物中的使用。巴瑞克替尼磷酸盐a晶型、h晶型和i晶型可以在治疗自身免疫疾病、炎性疾病或癌症的药物中应用,并且具有较好的生物利用度,同时提供的定性定量信息,对进一步研究此类固体药物的疗效具有重要的意义。附图说明图1为本发明提供的巴瑞克替尼磷酸盐a晶型的xrpd图谱。图2为本发明提供的巴瑞克替尼磷酸盐a晶型的高温稳定性的xrpd图谱(a:5天,b:10天)。图3为本发明提供的巴瑞克替尼磷酸盐a晶型的高湿稳定性的xrpd图谱(a:5天,b:10天)。图4为本发明提供的巴瑞克替尼磷酸盐a晶型的光照稳定性的xrpd图谱(a:5天,b:10天)。图5为本发明提供的巴瑞克替尼磷酸盐a晶型与现有的晶型x巴瑞克替尼磷酸盐在水中的溶解度曲线。图6为本发明提供的巴瑞克替尼磷酸盐h晶型的xrpd图谱。图7为本发明提供的巴瑞克替尼磷酸盐h晶型的高温稳定性的xrpd图谱(a:5天,b:10天)。图8为本发明提供的巴瑞克替尼磷酸盐h晶型的高湿稳定性的xrpd图谱(a:5天,b:10天)。图9为本发明提供的巴瑞克替尼磷酸盐h晶型的光照稳定性的xrpd图谱(a:5天,b:10天)。图10为本发明提供的巴瑞克替尼磷酸盐h晶型与现有的晶型x巴瑞克替尼磷酸盐在水中的溶解度曲线图11为本发明提供的巴瑞克替尼磷酸盐i晶型的xrpd图谱。图12为本发明提供的巴瑞克替尼磷酸盐i晶型的高温稳定性的xrpd图谱(a:5天,b:10天)。图13为本发明提供的巴瑞克替尼磷酸盐i晶型的高湿稳定性的xrpd图谱(a:5天,b:10天)。图14为本发明提供的巴瑞克替尼磷酸盐i晶型的光照稳定性的xrpd图谱(a:5天,b:10天)。图15为本发明提供的巴瑞克替尼磷酸盐i晶型与现有的晶型x巴瑞克替尼磷酸盐在水中的溶解度曲线图16为现有的晶型x巴瑞克替尼磷酸盐的xrpd图谱。图17为现有的晶型x巴瑞克替尼磷酸盐的高温稳定性的xrpd图谱(a:5天,b:10天)。图18为现有的晶型x巴瑞克替尼磷酸盐的高湿稳定性的xrpd图谱(a:5天,b:10天)。图19为现有的晶型x巴瑞克替尼磷酸盐的光照稳定性的xrpd图谱(a:5天,b:10天)。具体实施方式从下文的详细描述中,本发明的上述方面和本发明的其他方面将是明显的。实施例1至5巴瑞克替尼磷酸盐a晶型的制备称取1.0g巴瑞克替尼磷酸盐于样品瓶中,加入5mln,n二甲基甲酰胺使其完全溶解,随后在该溶解物中分别缓慢加入表1中的溶剂。待有固体从该溶解物和有机溶剂的混合物中析出后,再继续额外滴加该有机溶剂5ml。将所得到的混合物在室温下静置过夜,随后将静置过夜所得到的析出物过滤,并真空干燥,最后得到类白色固体,称量计算其收率,结果在表1中示出。过程中所使用的试剂均为分析纯。表1巴瑞克替尼a晶型的制备实施例溶剂种类溶剂量收率1乙醚20ml65%2异丙醚75ml61%3甲基叔丁基醚10ml61%4乙酸乙酯40ml60%5乙酸异丙酯50ml66%实施例6通过xrpd图来表征巴瑞克替尼磷酸盐a晶型x射线粉末衍射(xrpd)图谱的测量,使用rigakuultimaiv型号组合式多功能x射线衍射仪进行,具体采集信息如下:cu阳极(40kv,40ma),扫描速度20°/分钟、扫描范围(2θ范围)3~45°、扫描步长0.02、狭缝宽度0.01。采用载玻片直接在测试板压制对样品进行处理。其后的xrpd图谱均采用类似的测量方法。测定根据实施例1所述方法制备的巴瑞克替尼磷酸盐a晶型的xrpd图谱,在2θ=8.38,8.88,10.48,13.00,15.06,16.22,16.82,17.76,19.18,19.70,21.04,22.22,22.68,24.40,24.94,26.48,27.40处有衍射峰,如图1所示。其中2θ值误差范围为±0.2。经检测,2θ值误差范围也可以为±0.15。根据实施例2-5所述方法制备的巴瑞克替尼磷酸盐a晶型,其xrpd图谱与附图1所示图谱基本相同。本领域技术人员应理解,这些衍射峰不代表巴瑞克替尼磷酸盐a晶型所显示衍射峰的详尽情况。x射线粉末衍射图的2θ值是可以随着机器以及随着样品制备中的变化和批次间变化而轻微变化,所引用的值不视为绝对值。还应理解的是,峰的相对强度可能随取向效应而变,因此本发明所含的xrpd迹线中所示的强度是示例性的,并不用于绝对比较。实施例7巴瑞克替尼磷酸盐a晶型的高温稳定性考察取适量巴瑞克替尼磷酸盐a晶型置于60℃烘箱内,5天和10天后将该样品取出进行xrpd测试(如图2所示),以考察巴瑞克替尼磷酸盐a晶型对温度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐a晶型在高温条件下稳定。实施例8巴瑞克替尼磷酸盐a晶型的高湿稳定性考察取适量巴瑞克替尼磷酸盐a晶型置于92.5%湿度条件下,5天和10天后将该样品取出进行xrpd测试(如图3所示),以考察巴瑞克替尼磷酸盐a晶型对湿度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐a晶型在高湿条件下稳定。实施例9巴瑞克替尼磷酸盐a晶型的光照稳定性考察取适量巴瑞克替尼磷酸盐a晶型置于4500lux的光照强度下,5天和10天后将该样品取出进行xrpd测试(如图4所示),以考察巴瑞克替尼磷酸盐a晶型对光照的晶型稳定性。结果表明,巴瑞克替尼磷酸盐a晶型在光照条件下稳定。实施例10巴瑞克替尼磷酸盐a晶型与晶型x巴瑞克替尼磷酸盐的溶解度对比在35℃下,分别测定实施例1所得的巴瑞克替尼磷酸盐a晶型和比较例1所得的晶型x巴瑞克替尼磷酸盐在不同时间点的水溶解度,溶解度曲线如图5所示,结果表明,巴瑞克替尼磷酸盐a晶型在水中的溶解度为4.2mg/ml左右,并且在测定时间内维持于稳定的水平。晶型x巴瑞克替尼磷酸盐在水中的溶解度为2.5mg/ml左右。巴瑞克替尼磷酸盐a晶型在水中的溶解度大于晶型x巴瑞克替尼磷酸盐在水中的溶解度。实施例11巴瑞克替尼磷酸盐h晶型的制备称取1.0g巴瑞克替尼磷酸盐于样品瓶中,加入5mln,n二甲基甲酰胺使其完全溶解,随后在该溶解物中加入约3℃的乙酸乙酯20ml,立刻有固体从该溶解物和有机溶剂的混合物中析出,保持3℃下搅拌1小时。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例12巴瑞克替尼磷酸盐h晶型的制备称取1.0g巴瑞克替尼磷酸盐于样品瓶中,加入5mln,n二甲基甲酰胺使其完全溶解,随后在该溶解物中加入约-1℃的乙酸乙酯20ml,立刻有固体从该溶解物和有机溶剂的混合物中析出,保持-1℃下搅拌1小时。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例13巴瑞克替尼磷酸盐h晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入25ml乙腈和8ml乙醇的混合溶剂,随后将温度升至80℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有3.375mmol磷酸的乙醇溶液,在80℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液置于室温下冷却(通入25℃的降温介质条件降温),使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例14巴瑞克替尼磷酸盐h晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入24ml乙腈和8ml乙醇的混合溶剂,随后将温度升至85℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有3.75mmol磷酸的乙醇溶液,在85℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液置于室温下冷却(通入30℃的降温介质条件降温),使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例15巴瑞克替尼磷酸盐h晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入25ml乙腈和5ml乙醇的混合溶剂,随后将温度升至65℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有4.0mmol磷酸的乙醇溶液,在65℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液置于室温下冷却(通入20℃的降温介质条件降温),使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例16通过xrpd图来表征巴瑞克替尼磷酸盐h晶型以与实施例6相同的方法,测定根据实施例11所述方法制备的巴瑞克替尼磷酸盐h晶型的xrpd图谱,在2θ=7.94、12.401、13.539、14.278、15.957、16.779、17.52、19.403、20.282、22.259、24.617、26.04、27.144处有衍射峰,如图6所示。其中2θ值误差范围为±0.2。经检测,2θ值误差范围也可以为±0.15。根据实施例12-15所述方法制备的巴瑞克替尼磷酸盐h晶型,其xrpd图谱与附图6所示图谱基本相同。本领域技术人员应理解,这些衍射峰不代表巴瑞克替尼磷酸盐h晶型所显示衍射峰的详尽情况。x射线粉末衍射图的2θ值是可以随着机器以及随着样品制备中的变化和批次间变化而轻微变化,所引用的值不视为绝对值。还应理解的是,峰的相对强度可能随取向效应而变,因此本发明所含的xrpd迹线中所示的强度是示例性的,并不用于绝对比较。实施例17巴瑞克替尼磷酸盐h晶型的高温稳定性考察取适量巴瑞克替尼磷酸盐h晶型置于60℃烘箱内,5天和10天后将该样品取出进行xrpd测试(如图7所示),以考察巴瑞克替尼磷酸盐h晶型对温度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐h晶型在高温条件下稳定。实施例18巴瑞克替尼磷酸盐h晶型的高湿稳定性考察取适量巴瑞克替尼磷酸盐h晶型置于92.5%湿度条件下,5天和10天后将该样品取出进行xrpd测试(如图8所示),以考察巴瑞克替尼磷酸盐h晶型对湿度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐h晶型在高湿条件下稳定性一般。实施例19巴瑞克替尼磷酸盐h晶型的光照稳定性考察取适量巴瑞克替尼磷酸盐h晶型置于4500lux的光照强度下,5天和10天后将该样品取出进行xrpd测试(如图9所示),以考察巴瑞克替尼磷酸盐h晶型对光照的晶型稳定性。结果表明,巴瑞克替尼磷酸盐h晶型在光照条件下稳定。实施例20巴瑞克替尼磷酸盐h晶型与晶型x巴瑞克替尼磷酸盐的溶解度对比在35℃下,分别测定实施例11所得的巴瑞克替尼磷酸盐h晶型和比较例1所得的晶型x巴瑞克替尼磷酸盐在不同时间点的水溶解度,溶解度曲线如图10所示,结果表明,巴瑞克替尼磷酸盐h晶型在水中的溶解度为2.9mg/ml左右,并且在测定时间内维持于稳定的水平。晶型x巴瑞克替尼磷酸盐在水中的溶解度为2.5mg/ml左右。巴瑞克替尼磷酸盐h晶型在水中的溶解度大于晶型x巴瑞克替尼磷酸盐在水中的溶解度。实施例21巴瑞克替尼磷酸盐i晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入25ml乙腈和8ml乙醇的混合溶剂,随后将温度升至80℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有3.375mmol磷酸的乙醇溶液,在80℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液于90分钟内缓慢降至室温,使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例22巴瑞克替尼磷酸盐i晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入25ml乙腈和5ml乙醇的混合溶剂,随后将温度升至85℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有3.75mmol磷酸的乙醇溶液,在85℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液于100分钟内缓慢降至室温,使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例23巴瑞克替尼磷酸盐i晶型的制备称取1.0g巴瑞克替尼于样品瓶中,加入24ml乙腈和8ml乙醇的混合溶剂,随后将温度升至65℃使得巴瑞克替尼磷酸盐溶解。在2分钟内,向该溶解物中缓慢加入4ml含有4.0mmol磷酸的乙醇溶液,在65℃下将本步骤所得到的溶液继续搅拌2小时。随后将溶液于80分钟内缓慢降至室温,使固体析出。将所得到的析出物过滤,并真空干燥,最后得到类白色固体。过程中所使用的试剂均为分析纯。实施例24通过xrpd图来表征巴瑞克替尼磷酸盐i晶型以与实施例6相同的方法,测定根据实施例21所述方法制备的巴瑞克替尼磷酸盐h晶型的xrpd图谱,在2θ=8.818、10.442、12.901、14.901、16.076、16.88、17.54、18.677、19.12、19.44、20.596、21.699、22.422、23.181、24.539、25.779、27、28.461处有衍射峰,如图11所示。其中2θ值误差范围为±0.2。经检测,2θ值误差范围也可以为±0.15。根据实施例22-23所述方法制备的巴瑞克替尼磷酸盐h晶型,其xrpd图谱与附图11所示图谱基本相同。本领域技术人员应理解,这些衍射峰不代表巴瑞克替尼磷酸盐i晶型所显示衍射峰的详尽情况。x射线粉末衍射图的2θ值是可以随着机器以及随着样品制备中的变化和批次间变化而轻微变化,所引用的值不视为绝对值。还应理解的是,峰的相对强度可能随取向效应而变,因此本发明所含的xrpd迹线中所示的强度是示例性的,并不用于绝对比较。实施例25巴瑞克替尼磷酸盐i晶型的高温稳定性考察取适量巴瑞克替尼磷酸盐i晶型置于60℃烘箱内,5天和10天后将该样品取出进行xrpd测试(如图12所示),以考察巴瑞克替尼磷酸盐i晶型对温度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐i晶型在高温条件下稳定。实施例26巴瑞克替尼磷酸盐i晶型的高湿稳定性考察取适量巴瑞克替尼磷酸盐i晶型置于92.5%湿度条件下,5天和10天后将该样品取出进行xrpd测试(如图13所示),以考察巴瑞克替尼磷酸盐i晶型对湿度的晶型稳定性。结果表明,巴瑞克替尼磷酸盐i晶型在高湿条件下稳定性一般。实施例27巴瑞克替尼磷酸盐i晶型的光照稳定性考察取适量巴瑞克替尼磷酸盐i晶型置于4500lux的光照强度下,5天和10天后将该样品取出进行xrpd测试(如图14所示),以考察巴瑞克替尼磷酸盐i晶型对光照的晶型稳定性。结果表明,巴瑞克替尼磷酸盐i晶型在光照条件下稳定。实施例28巴瑞克替尼磷酸盐i晶型与晶型x巴瑞克替尼磷酸盐的溶解度对比在35℃下,分别测定实施例21所得的巴瑞克替尼磷酸盐i晶型和比较例1所得的晶型x巴瑞克替尼磷酸盐在不同时间点的水溶解度,溶解度曲线如图15所示,结果表明,巴瑞克替尼磷酸盐i晶型在水中的溶解度为3.6mg/ml左右,并且在测定时间内维持于稳定的水平。晶型x巴瑞克替尼磷酸盐在水中的溶解度为2.5mg/ml左右。巴瑞克替尼磷酸盐i晶型在水中的溶解度大于晶型x巴瑞克替尼磷酸盐在水中的溶解度。比较例1通过xrpd图来表征晶型x巴瑞克替尼磷酸盐根据中国发明专利cn102026999(wo2009/114512)中所公开的方法制备晶型x巴瑞克替尼磷酸盐。经xrpd测定,图谱如图16所示。所得到的固体熔点为188℃,和专利公开类似。比较例2晶型x巴瑞克替尼磷酸盐高温稳定性考察取适量晶型x巴瑞克替尼磷酸盐置于60℃烘箱内,5天和10天后将该样品取出进行xrpd测试(如图17所示),以考察晶型x巴瑞克替尼磷酸盐对温度的晶型稳定性。结果表明,晶型x巴瑞克替尼磷酸盐在高温条件下稳定。比较例3晶型x巴瑞克替尼磷酸盐高湿稳定性考察取适量晶型x巴瑞克替尼磷酸盐置于92.5%的湿度条件下,5天和10天后将该样品取出进行xrpd测试(如图18所示),以考察晶型x巴瑞克替尼磷酸盐对湿度的晶型稳定性。结果表明,晶型x巴瑞克替尼磷酸盐在高湿条件下稳定。比较例4晶型x巴瑞克替尼磷酸盐光照稳定性考察取适量晶型x巴瑞克替尼磷酸盐置于4500lux的光照强度下,5天和10天后将该样品取出进行xrpd测试(如图19所示),以考察晶型x巴瑞克替尼磷酸盐对光照的晶型稳定性。结果表明,晶型x巴瑞克替尼磷酸盐在光照条件下稳定。综上所述,巴瑞克替尼磷酸盐a晶型、h晶型和i晶型在高温、高湿及光照条件下都能够保持稳定,并且溶解度优于现有晶型。如本领域技术人员已知的,提高溶解度能够提高药物的生物利用度,并且稳定的晶型在药物制剂的生产过程中也具有优势。由于巴瑞克替尼磷酸盐a晶型、h晶型和i晶型具有的稳定性,其在各种固态剂型的药物加工过程中能够保持稳定,能够确定最终获得的药物中的药物活性成分的晶型,也能够获得更高的生物利用度,不会发生因为晶型转变而带来的药效差异。本领域的技术人员应当明了,尽管为了举例说明的目的,本文描述了本发明的具体实施方式,但可以对其进行各种修改而不偏离本发明的精神和范围。因此,本发明的具体实施方式和实施例不应当视为限制本发明的范围。本发明仅受所附权利要求的限制。本申请中引用的所有文献均完整地并入本文作为参考。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1