在无胺和水性条件下通过金属配合物催化由甲酸生成氢的制作方法
本申请要求于2016年6月29日提交的美国临时专利申请序列no.62/356,144的权益。
本发明大体上涉及用于生成氢气的催化剂及其用途。
背景技术:
本发明的背景将涉及包括氢、甲酸和催化剂的一般主题。
氢
氢是周期表中的第一种化学元素,并且具有的原子质量大约为1.008,它是元素周期表中最轻的元素。单原子形式的氢(h)是宇宙中最丰富的化学物质,构成宇宙元素质量的约75%,占恒星、气体行星和分子云质量的很大一部分。在地球上,氢是第三丰富的元素,具有三种天然存在的同位素,氕(缩写为1h)、氘(缩写为2h或d)和氚(缩写为3h或t)。
最丰富的氢同位素(氕)具有一个质子、一个电子,并且无中子,占地球上氢的超过99.98%。氘是不常见的氢同位素,具有一个质子、一个电子和一个中子,仅占地球上氢的0.0156%。氚是非常罕见的氢同位素,在原子核中具有两个中子,并且据估计占氢原子的十亿分之一。基于中子的存在或不存在,这三种同位素之间的主要差异是质量。由于这种质量差异,当用氘(d)替代化学化合物中的氢(h)时,它提供了一种在化学反应中追踪氢的手段,用于分析化学反应机理和速度决定步骤(rsd)。
在正常条件下,元素氢作为同核双原子气体存在,其具有两个共价键合在一起的氢原子,由式h2表示。该气体无臭、无色、无味。氢气比空气轻,但也在空气中高度易燃。由于比空气轻,使得氢气成为需要空气浮力的应用的有吸引力的选择;然而,正如1937年兴登堡飞艇爆炸所证明的那样,在浮力应用中使用氢气可能会非常危险。虽然氢气的可燃性使其不适用于飞艇,但这种可燃性以及气体的相对丰度确实使其成为能源产生的有前途的选择。
氢是清洁能源载体,当氢在发动机或燃料电池中燃烧时产生的唯一排放物是水。值得注意的是,氢已被用作驱动火箭和航天飞机进入轨道的燃料,并且由于在氢的燃烧中无危险性或放射性物质产生,因而作为副产物产生的水足够纯净以供航天飞机上的工作人员使用。
利用氢的燃料电池是一种很有前途的技术,用于作为建筑物的能源来源使用,并且潜在地用于推进交通工具的电动机。通过氢和氧的结合可以产生电、热和水。经常将燃料电池与电池相比,因为两者都将由化学反应产生的能量转换成可用的电能。燃料电池的一个优点是只要供应燃料(氢气)就能产生电,而不会损失电荷。潜在地,氢气(h2)在可再生能源技术的未来中具有关键作用。
虽然氢在地球上是丰富的,但其通常与其他元素(诸如碳和氧)结合存在,并且为了用于燃料必须将其从化合物中分离。用于燃料用途的氢可以由许多且不同的工艺产生。水的电解是产生氢的一种手段,在其中使用电流将氢与氧分离,但往往是昂贵的。木炭的气化是产生氢的另一种手段,在其中使用非常高的温度从碳和水木炭组分中释放氢,但需要大的能量投入才能达到有效温度。氢气生产的工业手段是天然气转化工艺,在其中将甲烷(ch4)暴露于加热蒸汽以通过两阶段化学反应产生氢气和二氧化碳,然而,该过程需要天然气作为起始材料。
甲酸
甲酸(fa),即二氧化碳(co2)和氢的加合物,具有式hco2h,(可替代地hcooh)并且是生物质氧化的主要副产物。甲酸是一种非常容易获取的、经济、不易燃的商品液体,可以安全运输和储存,并且因此,它被认为是氢的潜在液体来源和储存材料。甲酸脱水仅产生可以很容易分离的气体——氢气(h2)和二氧化碳(co2)。
甲酸的体积氢容量达到53gh2/l,这相当于1.77kw·h/l的能量密度。因此,对于机动车和移动应用甲酸是一种有吸引力的高密度能源。通过将co2和h2结合到甲酸中以储存氢并除去co2,并分解甲酸以释放氢,可以实现可持续的且可逆的能量储存和利用循环。
已证实在温和条件下甲酸分解释放氢。然而,当考虑到适合工业上可行的应用的规模时,这种温和的方法面临挑战。在许多现有系统中,产生了痕量的一氧化碳(co)副产物,其可以使氢燃料电池中用于产生氢的甲酸或其他材料中毒,这可能导致用于产生氢的甲酸或其他材料的部分或完全失活。
因为由于水分、空气、酸或其他存在于大多数甲酸来源中的痕量杂质导致的材料不稳定,用于产生氢的甲酸或其他材料的寿命(如由转换数(ton)所表明)不令人满意或不足够长。目前的甲酸来源和用途不能提供促进快速反应速率、具有可接受的寿命(ton)或在有效氢生产系统所需的反应条件下保持稳定的甲酸。
催化剂
催化剂是增加化学反应速率而在该过程中不经历永久性化学变化的物质。启动化学反应需要一定量的活化能,与点火需要火花大致相同。催化剂通过提供进行反应的替代路径来起作用,该替代路径需要的活化能低于非催化反应路径。然后,与非催化反应中相比,在催化反应中进行反应必须克服的能垒更低,使得反应可以更快地进行。
催化剂具有两种通用类型——均相的和非均相的。均相催化剂是催化剂与至少一种反应物同相的催化剂。非均相催化剂处于与任何反应物都不同的相中。
均相催化剂典型地参与反应机理的一个或多个步骤,但不是最终产物的一部分。反应完成之后,催化剂在化学上保持不变。均相催化剂的实例包括添加到反应物水溶液的水性均相催化剂。在这种类型的催化中,催化剂与反应物相互作用形成中间体,该中间体比原始反应物反应更快。通过提供比不存在催化剂时需要更少活化能的反应途径,中间体增加了反应物之间可以发生的相互作用。
非均相催化剂典型地包括与液体或气体反应物一起的固体催化剂。这些催化剂通过使反应物彼此足够接近以增加相互作用速率而起作用。这通常发生在当一种或多种反应物吸附在催化剂表面上使得反应物足够接近以促进相互作用时。
非均相催化剂的实例是机动车中的催化转化器。一氧化碳是在汽油发动机中发生的燃烧反应的有毒副产物,并且可以与氧气自发地反应产生二氧化碳;然而,即使该反应是自发的,这个过程也很慢。催化转化器通过提供将气体吸引到表面的材料(通常是金属,诸如铂)来加速该过程。一旦一氧化碳和氧气紧密接近,该反应就可以足够快地进行以产生二氧化碳,从而减少离开机动车排气系统的一氧化碳的量。
氢气(h2)是一种清洁的燃烧燃料,其潜在地在可再生能源技术的未来中具有关键作用;然而,需要能够安全且有效地从化学底物中释放氢气而不会产生有害的副产物或将使催化剂失活的副产物的催化剂化合物。此外,需要具有令人满意且足够的寿命(通过转换数(ton)测量),在水分、空气、酸或杂质的存在下具有材料稳定性,促进快速反应速率,并且在有效的氢生产系统所需的反应条件下保持稳定的用于产生氢的甲酸或其他材料的来源和用途。
技术实现要素:
本发明提供了一类可以安全且有效地从化学底物中释放氢气而不产生有害的副产物或将使催化剂失活的副产物的催化剂化合物。本发明还满足了对具有令人满意且足够的寿命(通过转换数(ton)测量),在水分、空气、酸或杂质的存在下具有材料稳定性,促进快速反应速率,并且在有效的氢生产系统所需的反应条件下保持稳定的用于产生氢的甲酸或其他材料的来源和用途的需要。
本文描述了催化剂和使用该催化剂从甲酸产生氢的方法。如本文所述的一类催化剂包括下式的化合物:
或其盐,其中,l是中性配体或阴离子配体;m是金属或金属离子;n为0、1或2;x1是卤素、氢根或甲酸根离子,并且r1、r2、r3、r4、r5和r6各自独立地选自氢、取代或未取代的烷基和取代或未取代的芳基。化合物具有下式:
其中,x2是卤素。金属或金属离子包括钌(ru)或铁(fe)。r1、r2、r3、r4、r5和r6可以各自为氢。l也可以是取代的芳基(例如对伞花烃)。化合物具有以下结构:
如本文所述的催化剂包括下式的化合物:
或其盐,其中,l是中性配体或阴离子配体;m是金属或金属离子;n为0、1或2;x1是卤素、氢根或甲酸根离子,并且r1、r3、r4、r5和r6各自独立地选自氢、取代或未取代的烷基和取代或未取代的芳基,且r2选自氢、取代或未取代的烷基和取代或未取代的芳基、n(ch3)2、nh2、oh、ch3o、c2h5o、ch3、f、i、cf3、cn或no2。化合物具有下式:
其中,x2是卤素。金属或金属离子包括钌(ru)或铁(fe)。r1、r2、r3、r4、r5和r6可以各自为氢。l也可以是取代的芳基(例如对伞花烃)。化合物具有下式:
其中,l是中性配体或阴离子配体,r2是n(ch3)2、nh2、oh、ch3o、c2h5o、ch3、f、i、cf3、cn或no2。
本文还描述了由甲酸和/或甲酸盐/酯(甲酸根)产生氢的方法。由甲酸和/或甲酸盐/酯产生氢的方法包括使甲酸和/或甲酸盐/酯(甲酸根)与如本文所述的催化剂接触,其中,接触步骤产生氢。接触步骤产生一种或多种另外的气体。该一种或多种另外的气体可以包括二氧化碳。该一种或多种另外的气体基本上不含或不含一氧化碳。甲酸盐/酯包括甲酸钠、甲酸钾和/或甲酸锂。接触步骤在溶剂的存在下进行。
溶剂包括水性溶剂(例如水)或甲醇。溶剂基本上不含或不含有机溶剂。接触步骤基本上不含或不含胺类。接触步骤可以在室温下进行。用于本文方法的催化剂具有从约500至约2,000,000的转换数(ton)和/或从约5,000h-1至约100,000h-1的转换频率(tof)。
本文另外描述了产生甲酸和/或甲酸盐/酯的方法。产生甲酸和/或甲酸盐/酯的方法包括使二氧化碳和氢气与如本文所述的催化剂接触。接触步骤在与大气压相比增加的压力下进行,并且接触步骤产生甲酸和/或甲酸盐/酯。
本文还描述了用于发电的方法。如本文所述的用于发电的方法包括(a)提供包括甲酸和/或甲酸盐/酯的氢源、如本文所述的催化剂、反应室和发电的燃料电池;(b)将氢源(例如甲酸和/或甲酸盐/酯)和催化剂输送到反应室,其中,反应室中的催化反应使甲酸和/或甲酸盐/酯转化成氢和二氧化碳;以及(c)将转化的氢输送到燃料电池以发电。
在下面的描述和附图中阐明了一种或多种实施方式的细节。根据详细描述和附图以及根据权利要求,其他特征、目的和优点将是显而易见的。
附图说明
通过结合附图考虑以下的详细描述,将理解本发明的上述和其他目的和优点,其中,相同的附图标记始终表示相同的部分,并且其中:
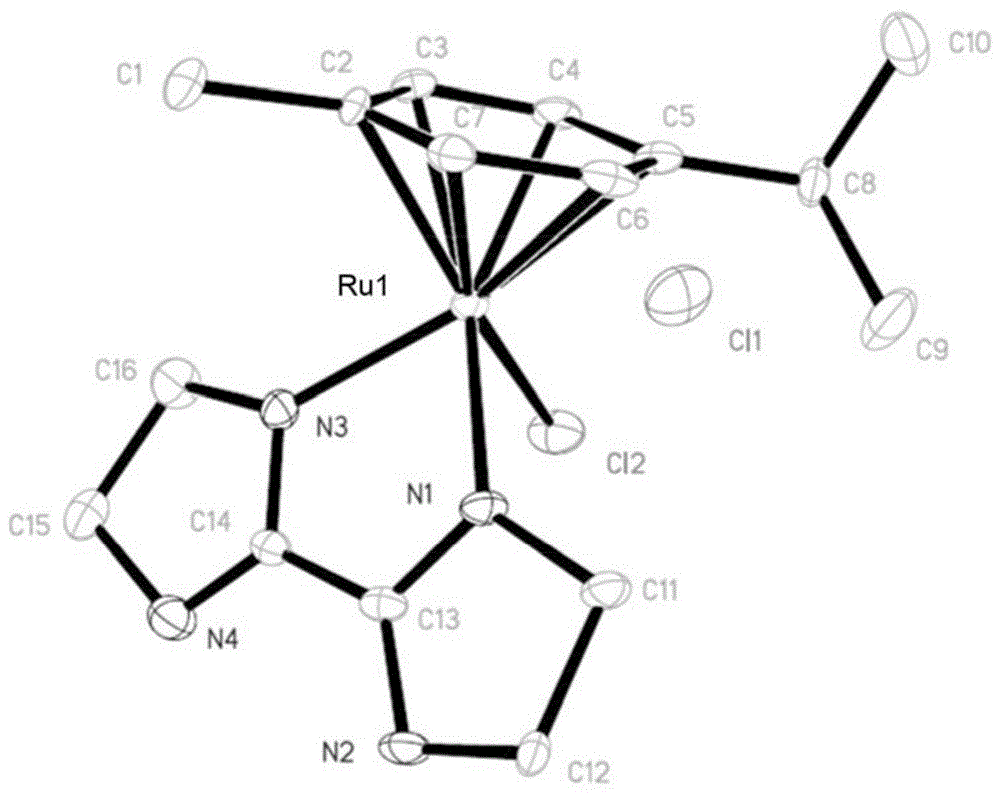
图1是化合物1/配合物1的x射线衍射晶体学结构。
图2a是示出了由配合物1催化的甲酸脱氢反应生成的气体混合物的组成分析的图。上图是co2/co气体标准的参照图,且下图是示出了co2检测的样品图。
图2b是示出了由配合物1催化的甲酸脱氢反应生成的气体混合物的组成分析的图。上图是h2气体标准的参照图,且下图是示出了h2检测的样品图。
图3a是在不同温度下在配合物1的存在下甲酸分解产生氢和二氧化碳的气体体积相对于时间的图。
图3b是使用配合物1分解甲酸的初始tof(转换频率)的阿利纽斯图(arrheniusplot)。
图4是配合物1在甲酸分解反应中的时间相关的性能的图,如ton(转换数)相对于时间所示。
图5a包含钌二聚体活化成氯配合物的自由能谱。
图5b示出了in1氯配合物的x射线结构相比于优化结构的键长参数。
图6a包含根据方案2中提出的机理的结构的相对自由能谱。
图6b示出了方案2的中间体和过渡化合物的结构。
图7包含氢转移过渡态和中间体的优化几何结构以及形成h2过渡态的优化几何结构的结构。
图8包含过渡态的优化结构和活化能垒。
图9a是用l3配体进行对位取代的结构。
图9b是对位取代的σ常数相对于平衡常数绘制的图。
虽然本发明易于接受各种修改和替代形式,但是其特定实施方式已经通过附图中的实例示出并在本文中详细描述。应当理解,本文对特定实施方式的描述并非旨在将本发明限制于所公开的特定形式,相反,其意图旨在涵盖落入如由所附权利要求限定的本发明的精神和范围内的所有修改、等同物和替代物。
具体实施方式
本发明提供了可以安全且有效地从化学底物中释放氢气而不产生有害的副产物或将使催化剂失活的副产物的催化剂化合物。本发明还满足了对具有令人满意且足够的寿命(通过转换数(ton)测量),在水分、空气、酸或杂质的存在下具有材料稳定性,促进快速反应速率,并在有效的氢生产系统所需的反应条件下保持稳定的用于产生氢的甲酸或其他材料的来源和用途的需要。
氢(h2)是清洁的燃烧燃料,其在可再生能源技术的未来中潜在地具有关键作用。本文描述了用作催化剂的化合物,以及使用所公开的催化剂从甲酸和/或甲酸盐/酯产生氢的方法。该方法包括使甲酸和/或甲酸盐/酯与如本文所述的催化剂接触,以及使用所公开的催化剂产生甲酸和/或甲酸盐/酯的方法和使用本文所述的催化剂发电的方法。在这些方法中,水或甲醇可用作溶剂而无需任何其他有机溶剂。
此外,反应不需要有机胺碱,且反应可在室温下进行。另外,甲酸到氢的转化在不生成大量一氧化碳或不生成任何一氧化碳的情况下进行。本文还提供了使用本文所述的催化剂由氢和二氧化碳产生甲酸和/或甲酸盐/酯的方法。本文还描述了通过使用本文所述的催化剂发电的方法。
化合物
本文所述的一类催化剂具有选自以下的杂环配体:
配体l1、l3、l5、l7和l8是可以在催化剂中使用的非芳香族配体。配体l2、l4、l6和l9是可以在催化剂中使用的芳香族配体。任选地,配体是l1或l3。
任选地,该类催化剂由式i:
及其盐表示。
在式i中,l是中性配体或阴离子配体。l可以是取代的芳基。l可以由以下结构表示:
在该结构中,ra、rb、rc、rd、re和rf各自独立地选自氢和取代或未取代的烷基。ra和rd是取代或未取代的烷基,并且rb、rc、re和rf是氢。l还可以是对伞花烃。
同样在式i中,m是金属或金属离子。该金属或金属离子可以是过渡金属。例如,该金属或金属离子可以是第4族的金属或金属离子、第5族的金属或金属离子、第6族的金属或金属离子、第7族的金属或金属离子、第8族的金属或金属离子、第9族的金属或金属离子、第10族的金属或金属离子或第11族的金属或金属离子。该金属或金属离子包括钌(ru)、铁(fe)、锇(os)、
另外,在式i中,n为0、1或2。
此外,在式i中,r1、r2、r3、r4、r5和r6各自独立地选自氢、取代或未取代的烷基和取代或未取代的芳基。任选地,r1、r2、r3、r4、r5和r6中的一个或多个是氢。任选地,r1、r2、r3、r4、r5和r6各自为氢。
此外,在式i中,x1是卤素、氢根或甲酸根离子。x1可以是氯、溴或氟。
式i可以由结构i-a表示:
在结构i-a中,l、m、n、r1、r2、r3、r4、r5、r6和x1是如上对式i所定义的。
同样在结构i-a中,x2是卤素。x2可以是氯、溴或氟。
举例来说,式i包括以下化合物:
任选地,该类催化剂由式ii:
及其盐表示。
在式ii中,l是中性配体或阴离子配体。l可以是取代的芳基。l可以用以下结构表示:
在该结构中,ra、rb、rc、rd、re和rf各自独立地选自氢和取代或未取代的烷基。ra和rd是取代或未取代的烷基,且rb、rc、re和rf是氢。l还可以是对伞花烃。
同样在式ii中,m是金属或金属离子。该金属或金属离子可以是过渡金属。例如,该金属或金属离子可以是第4族的金属或金属离子、第5族的金属或金属离子、第6族的金属或金属离子、第7族的金属或金属离子、第8族的金属或金属离子、第9族的金属或金属离子、第10族的金属或金属离子或第11族的金属或金属离子。该金属或金属离子包括钌(ru)、铁(fe)、锇(os)、
另外,在式ii中,n为0、1或2。
此外,在式ii中,r1、r3、r4、r5和r6各自独立地选自氢、取代或未取代的烷基和取代或未取代的芳基。任选地,r2选自氢、取代或未取代的烷基以及取代或未取代的芳基、n(ch3)2、nh2、oh、ch3o、c2h5o、ch3、f、i、cf3、cn或no2。r1、r2、r3、r4、r5和r6中的一个或多个是氢。任选地,r1、r2、r3、r4、r5和r6各自为氢。
此外,在式ii中,x1是卤素、氢根或甲酸根离子。x1可以是氯、溴或氟。
式ii可以由结构ii-a表示:
在结构ii-a中,l、m、n、r1、r2、r3、r4、r5、r6和x1是如上对式ii所定义的。
同样在结构ii-a中,x2是卤素。x2可以是氯、溴或氟。
举例来说,式ii包括以下结构ii-b:
在结构ii-b中,x1是甲酸根离子,m是钌,l是中性配体或阴离子配体。l可以是取代的芳基。l还可以是对伞花烃。r2可以是氢、取代或未取代的烷基、取代或未取代的芳基、给电子取代基:n(ch3)2、nh2、oh、ch3o、c2h5o或ch3,或吸电子取代基:f、i、cf3、cn或no2。任选地,r是n(ch3)2、ch3o、c2h5o或ch3。
如本文所用,术语烷基包括直链和支链一价取代基。实例包括甲基、乙基、异丙基、异丁基等。可用于本文所述化合物和方法的该基团的范围包括c1-c20烷基。可用于本文所述化合物和方法的这些基团的其他范围包括c1-c12烷基、c1-c6烷基和c1-c4烷基。
芳基分子包括,例如,包含一个或多个通过离域电子连接的典型地六个碳原子的平面组的环状烃,离域电子的数目与假设它们由交替的单和双共价键组成时相同。芳基分子的实例是苯。芳基分子还可包括另外的稠环,例如萘和蒽。除非另有说明,否则芳基分子可以附接在环上的任何位置。
本文使用的烷基和芳基分子可以是取代的或未取代的。如本文所用,术语取代包括将烷氧基、芳氧基、氨基、烷基、烯基、炔基、芳基、杂烷基、杂烯基、杂炔基、杂芳基、环烷基或杂环烷基基团加成到与烷基或芳基的主链附接的位置,例如,氢被这些分子中的一个替换。取代基团的实例包括但不限于羟基、卤素(例如f、br、cl或i)和羧基基团。相反,如本文所用,术语未取代表示烷基或芳基具有满额的氢,即与其饱和水平相当,没有取代,例如,线性癸烷(–(ch2)9–ch3)。
制备化合物的方法
本文所述的化合物可以以多种方式并使用各种合成方法制备。这些方法中的至少一些包括合成有机化学的使用。本文所述的化合物可由容易获得的起始材料制备。式i上的变化包括如对每种化合物所述的各种成分的添加、减少或移动。类似地,当分子中存在一个或多个手性中心时,包括所有可能的手性变体。另外,化合物合成可涉及各种化学基团的保护和去保护。可以确定保护和去保护的使用以及适当的保护基团的选择。
产生本文所述化合物的反应可在溶剂中进行,溶剂可通过有机合成来选择。在进行反应的条件例如温度和压力下,溶剂与起始材料(反应物)、中间体或产物可以是基本上不反应的。反应可以在一种溶剂或多于一种溶剂的混合物中进行。可以监测产物或中间体的形成。例如,可以通过光谱手段,诸如核磁共振光谱(例如,1h或13c)、红外光谱、分光光度法(例如,uv-可见光),或质谱,或通过色谱,诸如高效液相色谱(hplc)或薄层色谱来监测产物形成。
用于合成如本文所述的化合物的示例性方法在下文实施例1中提供。
使用的方法
本文提供了由甲酸和/或甲酸盐/酯产生氢的方法。产生氢的方法包括在催化剂的存在下使甲酸脱氢。方法包括使甲酸和/或甲酸盐/酯与如本文所述的催化剂接触。接触步骤产生氢和一种或多种另外的气体。
例如,接触步骤还可以产生二氧化碳(co2)。一种或多种另外的气体基本上不含一氧化碳(co)。如本文所用,术语基本上不含意为基于产生的气体或存在的气体的重量,该组分的产生量或存在量小于0.1%、小于0.01%、小于0.001%或小于0.0001%。一种或多种另外的气体不含一氧化碳(即,产生0%的一氧化碳或存在不可检测的水平的二氧化碳)。
甲酸盐/酯可以是甲酸盐。例如,甲酸盐可以是甲酸钠、甲酸钾、甲酸锂或其组合。
使甲酸和/或甲酸盐/酯与催化剂接触的步骤在溶剂的存在下进行。溶剂包括水性溶剂,诸如水。溶剂包括醇,诸如甲醇。溶剂可以是水性溶剂和醇的组合。溶剂基本上不含任何有机溶剂(即,基于溶剂的重量,在接触步骤中存在的有机溶剂的量小于0.1%、小于0.01%、小于0.001%或小于0.0001%)。例如,用于接触步骤的溶剂基本上不含二甲基亚砜(dmso)、甲苯或其他类似的有机溶剂。在一些实例中,溶剂不含有机溶剂(即,0%有机溶剂或不可检测水平的溶剂)。
接触步骤不含任何碱添加剂。例如,接触步骤不含胺类,诸如三甲胺或其他类似的胺。接触步骤可在室温下在环境条件下进行。
本文所述的催化剂可溶于水并且能够在不含有机溶剂和胺碱的条件下催化从甲酸选择性产生氢。这消除了与使用这些有机溶剂和胺碱相关的潜在风险。另外,与类似的甲酸/胺体系相比,本文所述的无胺方法具有更高的氢密度。
在本文所述的用于催化甲酸和/或甲酸盐/酯的条件下,可以选择性地控制氢的释放,仅产生氢和二氧化碳。不产生一氧化碳,也不会生成固体或重的残留物。所描述的由甲酸产生氢是定义明确的途径,具有几乎100%的选择性。
催化剂具有的转换数(ton)为从约500至约2,000,000(例如,从约1,000至约1,000,000、从约2,500至约500,000或从约5,000至约350,000)。转换数是一摩尔催化剂在失活前可转化的底物的摩尔数。催化剂越稳定,ton将越大。
催化剂具有的转换频率(tof)为从约5,000h-1至约100,000h-1(例如,从约6,000h-1至约50,000h-1或从约8,000h-1至约12,000h-1)。催化剂的转换频率表征其每单位时间的活性水平。tof是每小时由一摩尔活性位点转化成所需产物的总摩尔数。tof越大,催化剂活性越高。
本文还提供了由二氧化碳和氢产生甲酸和/或甲酸盐/酯的方法。产生甲酸和/或甲酸盐/酯的方法包括使二氧化碳和氢与如本文所述的催化剂接触以产生甲酸和/或甲酸盐/酯。接触步骤在与大气压相比增加的压力下进行。压力为从约15psi至约500psi(例如,从约25psi至约450psi、从约50psi至约400psi、从约100psi至约300psi或从约125psi至约250psi)。
本文还描述了用于发电的方法。用于发电的方法包括提供包括甲酸和/或甲酸盐/酯的氢源、如本文所述的催化剂、反应室和发电装置(例如,燃料电池);在甲酸反应室中使甲酸和/或甲酸盐/酯转化为氢和二氧化碳的条件下,将甲酸和/或甲酸盐/酯以及如本文所述的催化剂输送到反应室中;并将转化的氢输送到装置(例如燃料电池)。将氢输送到燃料电池,例如,提供燃料电池发电所需要的输入。
燃料电池与功率转换器协作以将通过燃料电池的运行产生的电力转换为适当的电流以输送到电池。转换的电力可被输送到电池,该电池可以是向待供电的电力装置供电所必需的任何适当的电池。例如,电池可用于向电动机动车、便携式装置或氢加注站供电。因此,本文提供了通过使用甲酸和/或甲酸盐/酯来源以及如本文所述的催化剂、反应室和由甲酸和/或甲酸盐/酯转化为氢和二氧化碳支持的电力装置(例如,电池或燃料电池)来供电的电动机动车、便携式装置、氢加注站或其他设备。
实施例
以下实施例旨在进一步说明本文所述方法和组合物的某些方面,而并不旨在限制权利要求的范围。
一般实验程序:
除非另有说明,否则所有用金属配合物的实验均在干燥氩气氛下在手套箱中或使用标准schlenk技术进行。所有其他化学物质均可商购获得并按原样使用。对于氢和碳,1h和13cnmr谱分别在bruker400mhz或600mhz光谱仪上以400mhz和100mhz或600mhz和125mhz记录。在硅胶(200-300目)上进行柱色谱法。
使用配备有热导检测器(tcd)和火焰离子化检测器(fid)的techcompgc7890ⅱ获得气相色谱分析结果。在使用标准缓冲溶液校准后的具有玻璃电极的zdj-400dh多功能滴定仪上测量ph值。在finniganmat95系统上记录高分辨率质谱(hrms)数据。在flash2000元素分析仪上进行元素分析。使用bruker-axskappa-apexiiccd衍射仪收集x射线衍射数据。在实验室中合成n,n'-二亚胺配体。计算转换数(ton)和转换频率(tof)。
实施例1:化合物的合成
配合物1[ru(对伞花烃)(thbid)cl]cl的合成和表征:
方案1
在手套箱中,将干燥甲醇(30.0ml)添加到反应烧瓶中的二氯(对伞花烃)钌(ii)二聚体(0.20mmol)和2,2'-联-2-咪唑啉(0.40mmol)的混合物中,并在室温下搅拌过夜。然后在减压下除去甲醇。使用柱色谱法纯化配合物,使用ch2cl2/meoh(50:1-20:1)作为洗脱剂。产率:60重量%。从dmso和醚(1:20)的混合物中得到了配合物1的单晶。1hnmr(600mhz,dmso)δ8.61(s,1h),5.88(d,j=6.1hz,2h),5.60(d,j=6.0hz,2h),4.61–4.30(m,2h),3.90–3.77(m,4h),3.72(dd,j=24.6,12.6hz,2h),2.71(dt,j=13.8,6.9hz,1h),2.11(s,3h),1.16(d,j=6.9hz,6h).13c{1h}nmr(125mhz,dmso):δ(ppm)=159.09,102.37,99.52,83.08,80.98,55.39,46.45,31.15,22.49,18.97;m/z(esi+)409.1[m-cl]+;c16h24cl2run4的元素分析计算值:c43.24、h5.44、n12.61;实测值c42.79、h5.63、n12.73。
图1是在由方案1合成得到的配合物1的晶体上进行的x射线衍射晶体学的结构示意图。在配合物1中,对伞花烃(η6)与氯和2,2'-联-2-咪唑啉配体一起配位至ru中心,得到饱和的18电子阳离子配合物。配合物1在空气中本质上是稳定的并且是水溶性的(~30mg/ml)。
实施例2:甲酸分解
hco2h催化脱氢的一般程序:
甲酸(hco2h)分解实验在配备有回流冷凝器和磁力搅拌器的双颈隔膜入口圆底烧瓶中在适当的温度下进行。在使用前新鲜制备催化剂储备溶液。通过隔膜入口将水性催化剂储备溶液注入至包含5.0ml甲酸(hco2h)和/或甲酸钠(hcoona)水溶液的烧瓶中。通过adm2000流量计监测产生的气体,并且每2秒收集一次数据。通过水置换法确认h2的量。
气体组成分析
使用在线预校准的气相色谱仪(agilent7890b)测定甲酸分解实验的气体产物的特性(h2、co、ch4、co2)。气相色谱仪配备有以下两个通道:1)带有热导检测器的两个hayesepq柱和13x分子筛,使用氦(he)作为载气用于co2和co分析,以及2)带有热导检测器的hayesepq和13x分子筛,使用氩(ar)作为载气用于h2分析。在1分钟的初始反应时间之后,通过将产生的气体以15分钟增量直接吹扫到在线气相色谱仪来分析产生的气体。
通过间歇加入甲酸的甲酸脱氢程序:
将搅拌棒、水(5.0ml)和甲酸/甲酸钠(fa/sf)水溶液(5m,4:1)置于双颈反应烧瓶中。在90℃下预热溶液之后,将配合物1的水溶液(0.5ml,0.5μmol)注入至烧瓶中。基于消耗速率(0.1-0.3ml/hr),通过注射泵以恒定速率加入甲酸。
结果:
用在90℃的温度下水溶液中的各种fa/hcoona比率来检查甲酸(fa)的分解(见表1)。对于每个分解反应,使用2μmol的配合物1作为催化剂。在不存在催化剂的情况下无反应发生。每种条件重复至少两次,具有的误差小于5%。在最初的10分钟内测量生成的h2的体积。
基于产生的h2估算最大转换频率(tof)。对于条目1,使用了5.0ml的5.0m甲酸溶液。对于条目2,使用了5.0ml的5.0m甲酸钠/甲酸(1:1)溶液。对于条目3,使用了5.0ml的5.0m甲酸钠/甲酸(2:1)溶液。对于条目4,使用了5.0ml的2m甲酸钠/甲酸(5:1)溶液。对于条目5,使用了5.0ml的1m甲酸钠/甲酸(10:1)溶液。
仅使用甲酸溶液,获得了3750h-1的初始转换频率(tof)(见表1,条目1)。图2a和图2b示出了对表1的条目1收集的气体样品进行气相色谱的结果,在y轴上显示信号强度(201)并在x轴上显示保留时间(202)。在1分钟的初始反应时间之后,通过将产生的气体以15分钟增量直接吹扫到在线气相色谱仪对产生的气体进行分析。
图2a示出了使用氦载气对用于co2/co分析的标准气体观察到的保留时间(202)的参照图(203),以及对表1的条目1的样品气体观察到的保留时间的样品图(204)。在参照图(203)上,对气体标准观察到co2的峰在2.483处(209),且co的峰在7.180处(210)。对样品气体观察到峰在2.567(205)处,表明存在co2,而没有从样品气体中观察到co的峰。
图2b示出了使用氩载气对用于h2分析的标准气体观察到的峰的参照图(206),以及对表1的条目1的样品气体观察到的峰的样品图(207)。在参照图(206)上观察到h2(211)的峰位于0.767处。在样品图(207)上,观察到峰位于0.852(208)处,表明存在h2。基于使用标准气体的校准曲线测量出h2和co2的摩尔比为大约1:1,在样品中未检测到co。
当将一当量的hcoona(sf)混合在fa溶液中时,fa的分解速率加快至得到的tof为8125h-1,这几乎是不使用sf时的速率的两倍(见表1,条目2)。这些结果表明,额外的甲酸根(hcoo-)有利于水中fa的分解。当sf和fa的比率增加到5时,得到了12,000h-1的最大tof(见表1,条目4)。在所有情况下,通过气相色谱法确认产生的气体的成分为h2和co2的等摩尔混合物,而未检测到痕量co。
这些观察结果表明,由配合物1催化的fa分解是高度选择性的并且仅产生h2和co2,适用于燃料电池应用而无电极co中毒。
表1
实施例3:机理研究
通过测量由2μmol配合物1催化的甲酸分解的初始反应速率研究活化能(ea)。在60℃至90℃的温度范围内在5ml的5m甲酸和甲酸钠(1:1)溶液中测量反应速率。
在图3a中,绘制了在60℃(306)、70℃(305)、80℃(304)和90℃(303)下进行的反应的气体体积(301)和以分钟计的时间(302)。如图3a所示,温度升高使得在较短时间内产生较大的气体体积,表明初始反应速率随着反应温度的升高而增加。
图3b是在相同的反应条件下图3a中示出的分解的初始tof值的阿利纽斯图,证明了反应速率的温度依赖性。在该图上,将tof的对数相对于1/t作图,以确定活化能的趋势。在最初的10分钟内测定了tof。如图3b所示,通过阿利纽斯图估计的表观活化能(ea)为23kj/mol,其在由贵金属催化的fa脱氢反应的已知范围内。
为了确定催化过程中的速率决定步骤(rds),进行了动力学同位素效应(kie)研究。动力学同位素效应(kie)是当被研究的反应物中的原子中的一个被其同位素中的一个取代时化学反应速率的变化。对于本kie研究,底物和/或溶剂中的一个或多个氢(“常见的”氢,标记为h)被氘(“重”氢,标记为d)取代。对于研究中使用的每个反应,试剂包括2μmol的催化剂1和5ml的2.5m甲酸溶液。反应在90℃的温度下进行。在最初的10分钟内计算转换频率(tof)。使用以下等式计算kie:kie=tof(条目1)/tof(条目n)(n=2、3、4)。结果如表2所示。
表2
表2的条目1,在底物或溶剂中均无氘(d)取代,用作比较的基线。使用dcooh代替hcooh,反应速率降低至2.1±0.3的kie(见表2,条目2),但当使用hcood时,kie仅为1.4±0.2(见表2,条目3)。在d2o中用dcood观察到略微更大的kie为3.1±0.4(见表2,条目4)。这些结果表明,从甲酸根到ru的氢转移是催化循环中的速率决定步骤(rds)。
在图4中,当以恒定速率(等于甲酸消耗的速率)加入甲酸(hcooh)时,对反应的ton(401)和以小时计的时间(402)作图(403)。在这些受控的反应速率条件下,在大约35小时内达到了350,000的ton。
计算机计算:
使用gaussian09程序包进行所有计算。首先在无任何几何或对称约束下使用结合bsi基组(bsi表示对过渡金属采用sdd并对n、o、h和c采用全电子6-31g(d,p)的基组组合)的m06泛函优化所有物种的几何结构。m06是色散修正泛函,并且由于考虑到了弱相互作用,因而可以很好地估计反应能量。
通过smd(溶剂化模型密度)考虑水溶剂的溶剂化效应,smd是基于溶质的极化连续量子力学电荷密度的通用溶剂化模型。通过对频率的分析计算将每个稳定点分类为最低态或过渡态。进行相同水平上的irc(内禀反应坐标)计算以确认过渡态及其向前和向后的极小值之间的正确连接。通过在与smd溶剂模型相结合的m06/bsii(bsii表示对ru采用sdd并对n、o、h、c采用全电子6-311++g(2d,p)的基组组合)水平上进行单点计算来精修每个物种的能量。
进行计算以评估对通过配合物1的甲酸脱氢所提出的机理,并确定由实验观察到的反应的过渡态和能垒。所提出的机理如方案2所示。
方案2
在图5a中,计算的自由能谱示出了氯配合物in1(504)容易由二聚体配合物[rucl2(对伞花烃)]2(501)形成。单体(502)与2,2'-联-2-咪唑啉(503)相互作用产生氯配合物in1(504)具有的活化能为-21.6,而产生in-h2o(505)副产物具有更高的能垒1.8。
在图5b中,比较了in1氯配合物的优化几何结构和x射线衍射确定的几何结构。如图5b中的键长参数比较所示,氯配合物in1的优化结构(507)与氯配合物in1的x射线衍射确定的结构(506)一致。
图6中示出了方案2中所提出的机理的详细的自由能谱(600)。图6中的中间体和过渡态的相应结构和键参数在图7中示出,并且在图8中示出了s-过渡态的活化能垒。通过实验,在ph4.3下获得了最佳tof。
如图6a中所示,从氯配合物in1向氢配合物in4的氢转移(605)有两种可能的途径,包括途径#1(601)(涉及in2至in3至ts1)和途径#2(602)(涉及ts2至in5至in6至ts3至in7)。
如图6a的途径#1(601a)中所示,用水(经由611)置换in1中的氯配体得到中间体in2,其吸热7.8kcal/mol。甲酸根阴离子与ru中心以
从in3至ts1的活化能垒为20.3kcal/mol。然后定位了内界(inner-sphere)β-h消除过渡态s-ts1(图8),涉及ru-n配位键的断裂,但相应的活化能垒高于ts1的活化能垒(25.5kcal/mol相比于20.3kcal/mol)。图8示出了理论化的s-ts1键长几何结构。释放co2(经由641)得到稳定的氢化物中间体in4,其相对于in1为-1.9kcal/mol。
如图6a的途径#2(602a)中所示,中间体in1至过渡态ts2代表通过甲酸根阴离子(经由612)的nh部分的去质子,具有6.8kcal/mol的低活化能垒。通过实验可以观察到随后in5中间体失去氯配体和nh基团的质子(经由622)。
加入甲酸根(经由632)产生in6。甲酸根中间体in6到过渡态ts3失去了一个质子(经由642),并且相比in3具有更短的ru-o和ru-n配位键和更高的自由能。in6的理论键长几何结构可以在图7中查看。然而,正失去一个质子的外界氢转移过渡态ts3具有与ts1类似的自由能(20.2相比于20.3kcal/mol)。
根据结构参数,ts3是早期过渡态,其中ru-h键为
图6b示出了方案2中途径#1(601b)和途径#2(602b)的中间体(in)和过渡态(ts)的结构。经由如图6a中所示的相同的路径,两个途径都以in1开始并以in4结束。途径#1(601a)包括中间体和过渡态in1-in2-in3-ts1-in4(经由611-621-631-641)。in2、in3和ts1的结构如图6b(601b)中所示。
途径#2(602a)包括中间体和过渡态in1-ts2-in5-in6-ts3-ts7-in4(经由612-622-632-642-652-662)。ts2、in5、in6、ts3和in7的结构在图6b(602b)中示出。基于图6a中所说明的可比较的活化能垒和实验观察结果,但不受理论束缚,对于实际反应中的氢转移阶段,途径#1和#2两者均可竞争性地发生。
如图6a中所示,为了氢化物中间体in4转变为in1而形成h2(606),选择了两个质子源,包括甲酸和具有一个水的水合质子。用甲酸(经由613-623)作为质子源的过渡态ts5具有的活化能垒比用水合质子(经由614-624)的过渡态ts4具有的活化能垒高13.7kcal/mol。
根据图7中的结构参数可以看出,ts5具有比ts4更应变的结构,特别是ts5中的ru-h键
对于具有过渡态s-ts2和s-ts3的另一h2形成途径(见图8),分别为29.9kcal/mol和17.5kcal/mol的较高的活化能垒排除了该反应途径发生的可能性。
还研究了不涉及金属中心的机理(图8中的s-ts4),表明甲酸根阴离子的氢直接与nh基团的质子反应产生h2和co2,但45.3kcal/mol的活化能垒非常高。总的来说,外界氢转移阶段是速率决定步骤,并且经由可比较的途径——途径#1(601a)或途径#2(602a)发生,这由本文提供的实验结果支持。h2(606)的形成经由用通过一个水连接的水合质子作为质子源(经由604)的ts4发生。
总之,确定了当与ru配合时n,n'-二亚胺配体显示出良好的活性,分别得到12,000h-1和350,000的高tof和ton。进行了实验研究和dft研究以阐明fa/sf分解的合理机理。在ph为4.3时,n,n'-二亚胺配体的一个nh基团的去质子化可得到关键的氢转移步骤的两个竞争途径。
实施例4:l3配体
通过修饰配体以增强活性进一步研究了l3配体。用一系列给电子取代基和吸电子取代基取代r2基团,并观察与每个取代相关的活化能。
图9a示出了具有位于吡啶环对位的r2(902)和位于x1位的甲酸根离子(903)的结构ii-b(901)。对于图9a中的ru-l3配合物,结构ii-b具有钌金属中心和l3配体。l3配体具有的吉布斯(gibbs)能为
在吡啶环的对位(p-)r2(902)中引入一系列吸电子取代基和给电子取代基。图9b中的图示出了相对于反应平衡常数(905)的对数绘制的取代基的σ常数(904)。给电子取代基是a-f,且吸电子取代基是g-k。
表3示出了在吡啶环的对位r2取代的给电子基团a-f和吸电子基团g-k以及与括号中的基团相关的吉布斯能。
表3
当r2基团是给电子基团时,观察到活化能
所附权利要求的化合物和方法的范围不受本文所述的旨在说明权利要求的一些方面的特定化合物和方法的限制,并且在功能上等同的任何化合物和方法都在本公开的范围内。除了本文示出的和描述的那些之外,化合物和方法的各种修改旨在落入所附权利要求的范围内。此外,虽然仅具体描述了这些化合物和方法的某些代表性化合物、方法和方面,但其他化合物和方法也旨在落入所附权利要求的范围内。因此,本文可以明确地提及步骤、元素、组分或成分的组合;然而,即使没有明确说明,也包括步骤、元素、组分和成分的所有其他组合。
- 还没有人留言评论。精彩留言会获得点赞!