一种盐酸氨溴索有关物质的合成方法与流程
本发明涉及有机合成
技术领域:
,具体涉及一种盐酸氨溴索有关物质的合成方法。
背景技术:
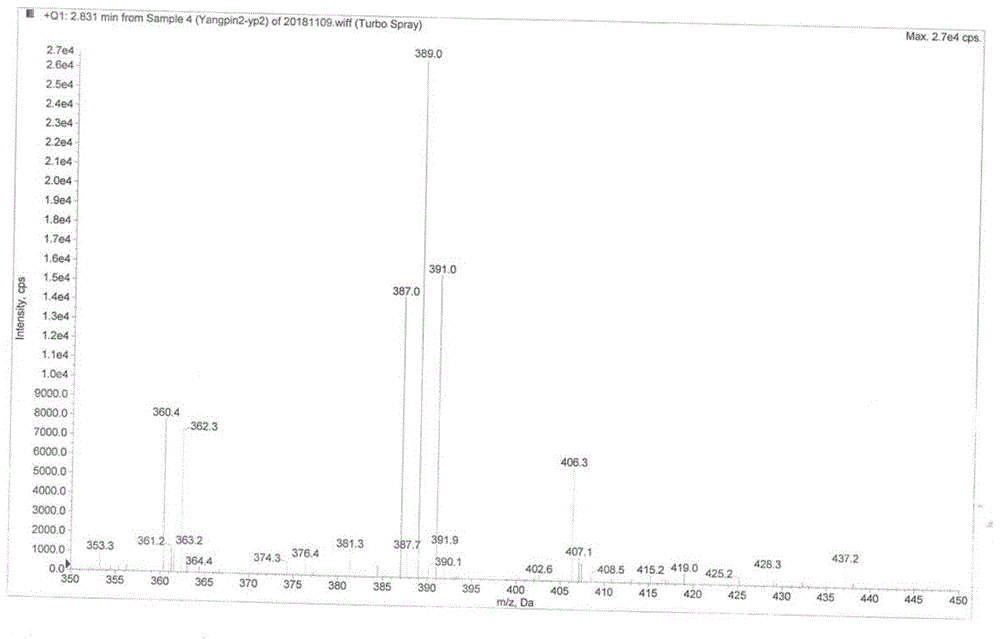
:盐酸氨溴索是一种作用很强的祛痰药,临床上常用于伴有痰液分泌不正常及排痰功能不良的急性、慢性肺部疾病,如慢性支气管炎急性加重、喘息型支气管炎及支气管哮喘的祛痰治疗;手术后肺部并发症的预防性治疗;早产儿及新生儿的婴儿呼吸窘迫综合症的治疗。盐酸氨溴索注射液已在国内外临床应用多年,其疗效确切、质量稳定,已获得了市场和消费者的普遍认可,临床用量大,尤其适合于危重、术后、禁食及其它不适合于口服的患者,并已进入我国各地医疗保险和工伤保险药品目录中。盐酸氨溴索的化学名为反式-4-[(2-氨基3.5-二溴苄基)氨基]环已醇盐酸盐,由德国勃林格殷格翰大药厂研究开发,80年代初在德国上市,商品名为“沐舒坦”,规格为2ml:15mg。盐酸氨溴索葡萄糖注射液在生产过程中会产生降解杂质4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇,该杂质在制剂灭菌过程中会显著生成。中国药科大学李园园采用超高效液相-离子阱-飞行时间串联质谱对该杂质进行了分离制备,结构表征,确定了其化学结构。目前,关于该杂质的合成及含量标定方法未见报道。鉴于该杂质对控制盐酸氨溴索产品质量至关重要,而其可作为本领域技术人员使用的标准品的制备方法和质量检测方法尚不可获得,故该杂质标准品的获得对有效控制盐酸氨溴索原料及其制剂质量有着重要的意义。文献《中国新药杂志》2006年第15卷第9期,page708-709,公开了氨溴索反式杂质6,8-二溴-3-(反式-4-羟基环己基)-1,2,3,4-四氢喹唑啉的详细制备过程。将氨溴索用甲醇和水(体积比:甲醇/水=3:1)溶解完全,缓慢滴入37%甲醛溶液,室温下反应10h。将反应液浓缩至干,残渣用石油醚-乙酸乙酯(体积比=2:1)的溶液重结晶得到产品。另外报道了采用将氨溴索原料药粗品用氯仿-丙酮(体积比=6:1)的溶液过柱洗脱从而得到产品。经过多次实验,我们发现该方法合成副产物较多,用石油醚-乙酸乙酯(体积比=2:1)分离纯化极为困难,收率较低,并且得到的产品纯度很低,无法满足作为药品杂质标准品的质量要求。gb1131164a和synthesisofmetabolitesofbisolvon.j.keck,liebigsann.chem.707,107-111(1967)也同样报道了氨溴索反式杂质6,8-二溴-3-(反式-4-羟基环己基)-1,2,3,4-四氢喹唑啉的制备过程,具体操作是将氨溴索用甲醇溶解,加入40%的甲醛,加热升温至回流反应2h,然后冷却至室温过滤,滤饼用甲醇/乙醚重结晶。但是研究中发现,合成收率较低,最高35%,产品分离纯化采用甲醇/乙醚重结晶,纯度较低,最高纯度不到95%,难以作为药品杂质标准品。由于上述路线制备得到的氨溴索反式杂质6,8-二溴-3-(反式-4-羟基环己基)-1,2,3,4-四氢喹唑啉纯度较低,因此难以满足作为药品杂质标准品的要求。技术实现要素:本发明所要解决的技术问题在于提供一种产物收率高、纯度高且工艺操作简单易行的盐酸氨溴索有关物质的合成方法,所制产物能够满足作为药品杂质标准品的要求。本发明所要解决的技术问题采用以下的技术方案来实现:一种盐酸氨溴索有关物质的合成方法,以盐酸氨溴索为原料,先与甲醛经环合反应合成4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐,再经氧化反应合成4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐。所述环合反应的环合体系由甲醛和溶剂组成,溶剂选自甲醇水溶液、乙醇水溶液中的一种。所述氧化反应的氧化体系由氧化剂和溶剂组成,氧化剂选自二氧化锰、硝酸铈铵、钯炭中的一种,溶剂选自二氯甲烷、甲醇、二氧六环、四氢呋喃、甲苯中的一种或多种。所述氧化反应的氧化体系由三氯化铁和酸性介质组成。所述酸性介质选自盐酸溶液、乙酸溶液中的一种。所述氧化反应的氧化体系由2价铜离子化合物、碱性介质和溶剂组成,溶剂选用dmso(二甲基亚砜)。所述2价铜离子化合物选自氯化铜、硝酸铜、溴化铜中的一种。所述碱性介质选用dbu(1,8-二氮杂二环十一碳-7-烯)。本发明的有益效果是:(1)本发明以盐酸氨溴索作为原料,与甲醛经环合反应制得4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐,环合反应的后处理过程中对溶剂进行了回收,降低成本的同时减小了废水处理难度;(2)本发明利用氧化反应由4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐合成4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐,分别采用了三种不同的氧化体系,最大程度地提高4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的转化率和4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的收率,使收率可达到90%以上,同时使纯度达到99%以上,满足作为药品杂质标准品的要求,从而利于控制盐酸氨溴索中杂质4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的含量。附图说明:图1为本发明产物4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的ms图谱;图2为本发明中间体4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的ms图谱;图3为本发明产物4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的1h-nmr图谱;图4为本发明中间体4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的1h-nmr图谱;图5为本发明产物4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的13c-nmr图谱;图6为本发明中间体4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐13c-nmr图谱。具体实施方式:为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例,进一步阐述本发明。实施例11、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成将盐酸氨溴索(8.3g,20mmol)、甲醇(60ml)、水(20ml),依次加入250ml圆底烧瓶中,缓缓加入37%的甲醛水溶液(15ml),25℃搅拌10h,tlc检测反应。反应结束后40℃减压回收甲醇,冷却析晶,抽滤,冷无水乙醇洗涤,50℃干燥,得到白色固体,收率93%,纯度98.6%。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、二氯甲烷(200ml)、硝酸铈铵(19.2g,35mmol)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例21、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、二氯甲烷(200ml)、二氧化锰(3g,35mmol)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例31、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、二氯甲烷(200ml)、10%钯炭(5g)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例41、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、六水三氯化铁(9.5g,35mmol)、1%盐酸溶液(30ml)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例51、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、dmso(10ml)、无水氯化铜(4.7g,35mmol)、dbu(2ml)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例61、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、dmso(10ml)、硝酸铜(6.6g,35mmol)、dbu(2ml)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。实施例71、4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐的合成同实施例1。2、4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐的合成将上一步产品(6g,14mmol)、dmso(10ml)、溴化铜(7.8g,35mmol)、dbu(2ml)依次加入250ml圆底烧瓶中,室温搅拌10h,tlc检测反应。反应结束后加水(100ml),分取水层,用5%碳酸钠溶液调节ph7-8,用乙酸乙酯(80ml*3)萃取,合并有机层,有机层用水(50ml*3)洗涤,无水硫酸钠干燥,减压回收乙酸乙酯至10ml,搅拌下加入浓盐酸(1ml),冷却析晶,抽滤,乙酸乙酯洗涤,50℃干燥,得到白色固体。对照例1合成方法同实施例4,只是不添加1%盐酸溶液。对照例2合成方法同实施例5,只是不添加dbu。对照例3合成方法同实施例6,只是不添加dbu。对照例4合成方法同实施例7,只是不添加dbu。对照例5合成方法同实施例5,只是将dbu替换成等摩尔量的氢氧化钠溶液。表1实施例1-8及对照例1-3第二步产品的收率和纯度组别氧化体系收率/%纯度/%实施例1硝酸铈铵+二氯甲烷5298.9实施例2二氧化锰+二氯甲烷6098.3实施例310%钯炭+二氯甲烷8198.7实施例4六水三氯化铁+1%盐酸溶液8599.8实施例5无水氯化铜+dbu+dmso9299.8实施例6硝酸铜+dbu+dmso8399.2实施例7溴化铜+dbu+dmso8998.5对照例1六水三氯化铁0/对照例2无水氯化铜+dmso0/对照例3硝酸铜+dmso0/对照例4溴化铜+dmso0/对照例5无水氯化铜+naoh+dmso7698.4化合物表征数据:4-(6,8-二溴-1,4-二氢喹唑啉-3(2h)-基)-环己醇盐酸盐:mp:226.8~229.3℃[4]。ir(kbr)υ(cm-1):3409.13,2912.56,2487.74,1598.35,1499.56,1068.34,866.90,816.66,669.29;1h-nmr(dmso-d6,400mhz)ppmδ:1.15-1.18(m,2h,ch2incyclohexane),1.58-1.60(m,2h,ch2incyclohexane),1.89-1.92(m,2h,ch2incyclohexane),2.06-2.18(m,2h,ch2incyclohexane),3.07(m,3h,n-ch),3.34-3.40(m,1h,o-ch),4.48brs,(3h,1/2ch2and1/2n-ch2-n),4.67(s,1h,1/2ch2),6.64(s,1h,nh),7.38(d,1h,j=2.0hz,chinar),7.67(d,1h,j=2.0hz,chinar),11.25(s,1h,oh);13c-nmr(dmso-d6,100mhz)ppmδ138.33,130.09,110.69,67.83,59.81,59.21,48.16,33.58,25.44,25.11;esi-ms(m/z):c14h18br2n2o,391.0[m+h]+.4-(6,8-二溴-3,4-二氢喹唑啉-3-基)-环己醇盐酸盐:mp:198.3~199.9℃。ir(kbr),υ,cm-1:3398.36,2922.35,1599.12,1497.35,1068.78,855.50,846.69;1h-nmr(dmso-d6,400mhz)ppmδ:1.19-1.29(m,2h,ch2incyclohexane),1.64-1.74(m,2h,ch2incyclohexane),1.83-1.93(m,4h,2×ch2incyclohexane),3.38-3.41n-ch),3.86-3.89(m,1h,o-ch),4.83(s,2h,ch2),7.42(s,1h,chinar),7.90(s,1h,chinar),8.25(s,1h,=ch),11.68(s,1h,oh);13c-nmr(dmso-d6,100mhz)ppmδ151.16,134.76,129.62,129.54,122.41,119.16,111.68,67.88,63.46,43.27,33.96,27.20;esi-ms(m/z):c14h16br2n2o,389.1[m+h]+.以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1