一种萘环-异噁唑型化合物及其制备方法与应用与流程
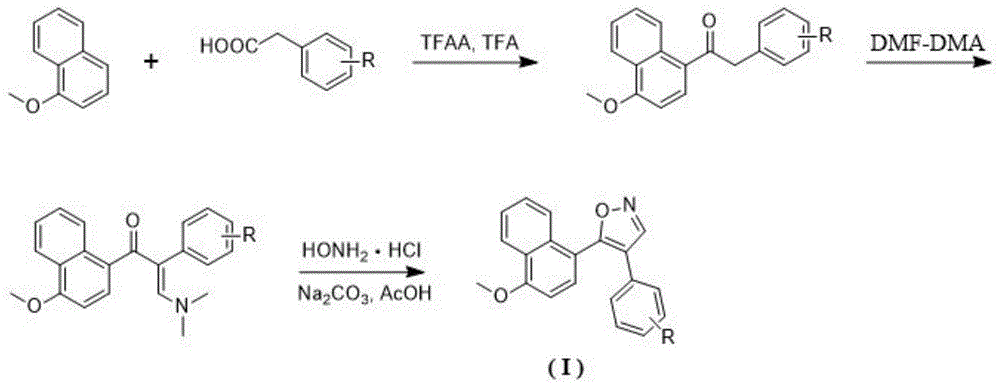
本发明属于化合物制备技术领域,具体涉及一种萘环-异噁唑型化合物及其制备方法和在制备微管蛋白抑制剂类抗肿瘤药物中的应用。
背景技术:
微管是真核细胞骨架的主要组成部分,由α和β微管蛋白以二聚体的形式堆积而成,具有不断的聚合和解聚的动力学特征。微管在真核细胞的复制、分裂、信号传导、维持细胞形状等多种细胞功能上具有非常重要的作用。由于微管在细胞分裂中的重要作用,现在已成为抗肿瘤药物研究的重要靶点之一。
异噁唑是一种重要的杂环型化合物,存在于许多天然产物和合成化合物之中。近年来,大量文献报道异噁唑及其衍生物具有抗真菌、抗微生物、抗炎、抗hiv、抗糖尿病、抗肿瘤等多种生物活性。另一方面,萘环也是一种重要的药物设计的药效团,很多萘环型化合物具有较好的生物活性。
由于异噁唑和萘环衍生物具有较好的生物活性和潜在的药用价值,现已成为药物化学研究中十分活跃的研究领域,研究和开发萘环-异噁唑类微管蛋白抑制剂类抗肿瘤药物具有十分重要的意义。
技术实现要素:
有鉴于此,本发明的目的是针对现有技术中存在的问题,提供一种萘环-异噁唑型化合物及其制备方法,以及提供上述萘环-异噁唑型化合物在制备微管蛋白抑制剂类抗肿瘤药物上的应用。
为了实现上述目的,本发明采用如下技术方案:
一种萘环-异噁唑型化合物,所述化合物具有如式(i)所示结构:
其中,通式i中的r至少为氢、氟、氯、溴、碘、c1~c3烷氧基、c1~c3烷基、硝基、胺基、羟基中的一种。
一种萘环-异噁唑型化合物的制备方法,所述方法具体包括以下步骤:
步骤1:将1-甲氧基萘、取代苯乙酸和三氟乙酸酐加入反应瓶中,再加入三氟乙酸作为溶剂,反应液在室温下搅拌反应12-24小时,制得1-(4-甲氧基萘-1-基)-2-取代苯基乙烷-1-酮;
步骤2:将1-(4-甲氧基萘-1-基)-2-取代苯基乙烷-1-酮和dmf-dma混匀,加热回流反应12-24小时,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-取代苯基丙-2-烯-1-酮;
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-取代苯基丙-2-烯-1-酮、盐酸羟胺、碳酸钠置于反应瓶中,加入体积比为2:1的甲醇-水混合液做溶剂,并调节反应液的ph为4-5,60~70℃下反应8-24小时,制备得到通式(i)化合物。
优选的,所述步骤1中,1-甲氧基萘、取代苯乙酸与三氟乙酸酐的摩尔比例为(1-2):1:(1-2)。
优选的,所述步骤2中,1-(4-甲氧基萘-1-基)-2-取代苯基乙烷-1-酮与dmf-dma的摩尔比例为1:(1-10)。
优选的,所述步骤3中,(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-取代苯基丙-2-烯-1-酮、盐酸羟胺和碳酸钠的摩尔比例为1:(1-2):(1-2)。
另外,本发明还请求保护上述萘环-异噁唑型化合物在制备微管蛋白抑制剂类抗肿瘤药物中的应用。
与现有技术相比,本发明公开的一种萘环-异噁唑型化合物及其制备方法与应用,具有如下优点:
1、本发明提供的一种萘环-异噁唑型化合物的制备方法简单,合成路线较短,反应条件温和,对实验设备要求较低,适于工业化生产;
2、本发明公开提供的萘环-异噁唑型化合物具有良好的抑制微管蛋白聚合的活性和抑制肿瘤细胞增殖的作用,可以作为研究新型微管蛋白抑制剂类抗肿瘤药物的先导化合物。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1为本发明一种萘环-异噁唑型化合物的制备流程图。
图2为本发明实施例提供的一种萘环-异噁唑型化合物对微管蛋白聚合影响图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为更好地理解本发明,下面通过以下实施例对本发明作进一步具体的阐述,但不可理解为对本发明的限定,对于本领域的技术人员根据上述发明内容所作的一些非本质的改进与调整,也视为落在本发明的保护范围内。
本发明公开了一种萘环-异噁唑型化合物,具有如式(i)所示结构通式:
其中,通式i中的r至少为氢、氟、氯、溴、碘、c1~c3烷氧基、c1~c3烷基、硝基、胺基、羟基中的一种。
如附图1所示,本发明还提供制备上述通式i的萘环-异噁唑型化合物的方法,该方法包括下列步骤:
步骤1:将1-甲氧基萘、取代苯乙酸和三氟乙酸酐加入反应瓶中,摩尔比为(1-2):1:(1-2),再加入三氟乙酸作为溶剂,反应液在室温下搅拌反应12-24小时,制得1-(4-甲氧基萘-1-基)-2-取代苯基乙烷-1-酮;
步骤2:将1-(4-甲氧基萘-1-基)-2-取代苯基乙烷-1-酮和dmf-dma混匀,摩尔比例为1:(1-10),回流反应12-24小时,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-取代苯基丙-2-烯-1-酮;
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-取代苯基丙-2-烯-1-酮、盐酸羟胺、碳酸钠置于反应瓶中,摩尔比例为1:1-2:1-2,加入甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应8-24小时,制备得到通式(i)化合物。
为了进一步说明本发明公开的技术方案,发明人还进行了如下实施例:
实施例一:
一种5-(4-甲氧基萘-1-基)-4-(3,4,5-三甲氧基苯基)异噁唑(1)的制备
化合物1的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3,4,5-三甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3,4,5-三甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3,4,5-三甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3,4,5-三甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3,4,5-三甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物1。
其中化合物1为黄色固体,收率为74%;且化合物1的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.47(s,6h),3.77(s,3h),4.05(s,3h),6.39(s,2h),6.86(d,1h,j=8.0hz),7.41-7.46(m,1h),7.47-7.51(m,1h),7.54(d,1h,j=8.0hz),7.63(d,1h,j=8.4hz),8.31(d,1h,j=7.6hz),8.63(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.9,55.9,61.0,103.3,104.5,117.6,117.8,122.5,125.1,125.3,125.7,125.9,127.8,129.7,132.1,137.4,150.2,153.4,157.5,164.7;
hrms(esi)calcdfor[m+h]+c23h22no5+:392.14925found392.14603.
以下实施例的制备方法与实施例一类似。
实施例二:
一种5-(4-甲氧基萘-1-基)-4-(4-甲氧基苯基)异噁唑(2)的制备
化合物2的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物2。
其中化合物2为黄色油状物,收率为75%;且化合物2的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.71(s,3h),4.02(s,3h),6.71(d,2h,j=8.8hz),6.81(d,1h,j=8.0hz),7.12(d,2h,j=8.8hz),7.41-7.45(m,1h),7.47-7.51(m,2h),7.69(d,1h,j=8.8hz),8.33(d,1h,j=8.4hz),8.60(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.3,55.8,103.5,114.3,117.7,117.7,122.1,122.5,125.3,125.8,125.9,127.7,128.6,129.6,132.2,150.6,157.4,159.0,164.2;
hrms(esi)calcdfor[m+h]+c21h18no3+:332.12812found332.12601.
实施例三:
一种5-(4-甲氧基萘-1-基)-4-(4-甲基苯基)异噁唑(3)的制备
化合物3的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-甲基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-甲基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-甲基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物3。
其中化合物3为黄色油状物,收率为86%;且化合物3的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):2.13(s,3h),3.98(s,3h),6.95(d,2h,j=8.0hz),7.01(dd,1h,j=8.0hz,1.2hz),7.10(d,2h,j=8.0hz),7.40-7.50(m,3h),7.53(d,1h,j=8.0hz),8.21(d,1h,j=8.4hz),9.12(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):21.2,56.4,104.7,117.5,118.1,122.7,125.0,125.5,126.5,126.7,127.3,128.3,129.8,130.2,132.0,137.4,151.4,157.3,163.8;
hrms(esi)calcdfor[m+h]+c21h18no2+:316.13321found316.13113.
实施例四:
一种4-(4-氯苯基)-5-(4-甲氧基萘-1-基)异噁唑(4)的制备
化合物4的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-氯苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-氯苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-氯苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-氯苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-氯苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物4。
其中化合物4为褐色油状物,收率为74%;且化合物4的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.04(s,3h),6.83(d,1h,j=8.0hz),7.11(d,2h,j=8.8hz),7.17(d,2h,j=8.8hz),7.41-7.45(m,1h),7.47-7.52(m,2h),7.64(d,1h,j=8.4hz),8.33(d,1h,j=8.0hz),8.60(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,103.5,117.0,117.1,122.6,125.0,125.8,126.0,127.9,128.3,128.6,129.1,129.6,132.0,133.4,150.3,157.7,165.3;
hrms(esi)calcdfor[m+h]+c20h15clno2+:336.07858found336.07648.
实施例五:
一种4-(2-氯苯基)-5-(4-甲氧基萘-1-基)异噁唑(5)的制备
化合物5的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-氯苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(2-氯苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(2-氯苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-氯苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-氯苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物5。
其中化合物5为黄色油状物,收率为59%;且化合物5的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.98(s,3h),6.73(d,1h,j=8.0hz),6.99-7.00(m,2h),7.14-7.18(m,1h),7.38-7.49(m,4h),7.82-7.84(m,1h),8.28-8.30(m,1h),8.63(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.7,103.3,115.5,117.0,122.4,125.1,125.7,125.8,127.0,127.6,129.2,129.4,129.6,130.1,131.7,132.0,133.6,151.9,157.5,166.7;
hrms(esi)calcdfor[m+h]+c20h15clno2+:336.07858found336.07669.
实施例六:
一种5-(4-甲氧基萘-1-基)-4-(2-甲氧基苯基)异噁唑(6)的制备
化合物6的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(2-甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(2-甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物6。
其中化合物6为黄色油状物,收率为65%;且化合物6的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.65(s,3h),4.00(s,3h),6.69(dt,1h,j=8.0hz,0.8hz),6.77(d,1h,j=8.0hz),6.86(d,1h,j=8.4hz),6.94(dd,1h,j=8.0hz,1.2hz),7.18(dt,1h,j=8.0hz,1.6hz),7.39-7.49(m,3h),7.76(d,1h,j=8.4hz),8.30(d,1h,j=8.0hz),8.71(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.3,55.7,103.3,111.1,114.3,118.3,118.8,120.7,122.3,125.4,125.7,127.4,129.2,130.3,132.2,152.3,156.7,157.2,165.5;
hrms(esi)calcdfor[m+h]+c21h18no3+:332.12812found332.12589.
实施例七:
一种4-(3-氟-4-甲氧基苯基)-5-(4-甲氧基萘-1-基)异噁唑(7)的制备
化合物7的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3-氟-4-甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3-氟-4-甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3-氟-4-甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氟-4-甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氟-4-甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物7。
其中化合物7为黄色油状物,收率为77%;且化合物7的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.79(s,3h),4.03(s,3h),6.75(t,1h,j=8.8hz),6.83(d,1h,j=8.8hz),6.90-6.94(m,2h),7.40-7.45(m,1h),7.47-7.50(m,2h),7.63(d,1h,j=8.8hz),8.32(d,1h,j=8.8hz),8.58(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,56.2,103.5,113.6,115.0(d,1c,j=19.4hz),116.8,117.2,122.6,122.7(d,1c,j=7.1hz),123.3(d,1c,j=3.3hz),125.0,125.8(d,1c,j=11.5hz),126.0,127.8,129.6,132.0,147.0(d,1c,j=10.6hz),150.3,151.1(d,1c,j=244hz),157.6,164.7;
hrms(esi)calcdfor[m+h]+c21h17fno3+:350.11870found350.11618.
实施例八:
一种5-(4-甲氧基萘-1-基)-4-(3-甲氧基苯基)异噁唑(8)的制备
化合物8的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3-甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3-甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3-甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物8。
其中化合物8为黄色油状物体,收率为72%;且化合物8的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.52(s,3h),4.05(s,3h),6.72-6.74(m,2h),6.80(d,1h,j=8.0hz),6.84(d,1h,j=8.0hz),7.10-7.14(m,1h),7.43-7.50(m,2h),7.51(d,1h,j=8.0hz),7.66(d,1h,j=8.4hz),8.32(d,1h,j=8.0hz),8.63(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.1,55.8,103.4,112.8,113.2,117.6,117.8,119.8,122.5,125.3,125.8,125.9,127.7,129.6,129.9,131.0,132.1,150.5,157.5,159.8,165.1;
hrms(esi)calcdfor[m+h]+c21h18no3+:332.12812found332.12585.
实施例九:
一种4-(3,4-二甲氧基苯基)-5-(4-甲氧基萘-1-基)异噁唑(9)的制备
化合物9的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3,4-二甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3,4-二甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3,4-二甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3,4-二甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3,4-二甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物9。
其中化合物9为黄色固体,收率为63%;且化合物9的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.36(s,3h),3.81(s,3h),4.04(s,3h),6.58(d,1h,j=2.0hz),6.72(d,1h,j=8.4hz),6.84-6.87(m,2h),7.40-7.44(m,1h),7.46-7.50(m,1h),7.53(d,1h,j=8.0hz),7.65(d,1h,j=8.4hz),8.31(d,1h,j=8.0hz),8.62(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.5,55.8,55.9,103.4,110.4,111.3,117.7,117.8,119.7,122.3,122.5,125.4,125.7,125.9,127.7,129.6,132.1,148.4,148.9,150.3,157.4,164.2;
hrms(esi)calcdfor[m+h]+c22h20no4+:362.13868found362.13586.
实施例十:
一种4-(4-乙氧基苯基)-5-(4-甲氧基萘-1-基)异噁唑(10)的制备
化合物10的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-乙氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-乙氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-乙氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-乙氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-乙氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物10。
其中化合物10为黄色固体,收率为68%;且化合物10的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):1.36(t,3h,j=6.8hz),3.92(q,2h,j=6.8hz),4.05(s,3h),6.71(d,2h,j=8.8hz),6.83(d,1h,j=8.0hz),7.10(d,2h,j=8.8hz),7.40-7.47(m,1h),7.49(d,2h,j=8.0hz),7.67(d,1h,j=8.4hz),8.32(d,1h,j=8.4hz),8.58(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):14.9,55.8,63.5,103.4,114.8,117.7,117.7,121.9,122.4,125.3,125.8,125.9,127.6,128.6,128.6,129.5,132.1,150.6,150.6,157.4,158.3,164.2;
hrms(esi)calcdfor[m+h]+c22h20no3+:346.14377found346.14105.
实施例十一:
一种5-(4-甲氧基萘-1-基)-4-(2-甲基苯基)异噁唑(11)的制备
化合物11的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-甲基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(2-甲基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(2-甲基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-甲基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-甲基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物11。
其中化合物11为黄色油状物,收率为92%;且化合物11的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):2.04(s,3h),3.98(s,3h),6.70(d,1h,j=8.0hz),7.09-7.19(m,4h),7.28(d,1h,j=8.0hz),7.46-7.51(m,2h),7.94-7.96(m,1h),8.28-8.30(m,1h),8.43(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):20.4,55.7,103.3,117.1,117.5,122.4,125.3,125.8,125.8,126.2,127.6,128.2,129.3,129.6,130.5,130.6,131.9,136.8,152.1,157.3,166.2;
hrms(esi)calcdfor[m+h]+c21h18no2+:316.13321found316.13095.
实施例十二:
一种4-(2-氟苯基)-5-(4-甲氧基萘-1-基)异噁唑(12)的制备
化合物12的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-氟苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(2-氟苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(2-氟苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-氟苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-氟苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物12。
其中化合物12为白色固体,收率为73%;且化合物12的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.04(s,3h),6.81(d,1h,j=8.0hz),6.83-6.87(m,1h),6.96(dt,1h,j=8.0hz,2.0hz),7.05-7.10(m,1h),7.15-7.21(m,1h),7.41-7.51(m,3h),7.69(d,1h,j=8.4hz),8.31(d,1h,j=8.0hz),8.68(d,1h,j=2.4hz);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,103.4,112.0,116.0(d,1c,j=21.6hz),117.3,117.7(d,1c,j=14.1hz),122.5,124.3(d,1c,j=3.5hz),125.1,125.8,125.9,127.7,129.4,129.5,130.1(d,1c,j=3.0hz),132.0,151.4(d,1c,j=6.6hz),157.5,158.6(d,1c,j=247hz),166.1;
hrms(esi)calcdfor[m+h]+c20h15fno2+:320.10813found320.10593.
实施例十三:
一种4-(苯并[d][1,3]二氧杂环戊烯-5-基)-5-(4-甲氧基萘-1-基)异噁唑(13)的制备
化合物13的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(苯并[d][1,3]二氧杂环戊烯-5-基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(苯并[d][1,3]二氧杂环戊烯-5-基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物13。
其中化合物13为黄色固体,收率为79%;且化合物13的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.05(s,3h),5.88(s,2h),6.63-6.67(m,2h),6.71(dd,1h,j=8.0hz,2.0hz),6.84(d,1h,j=8.0hz),7.42-7.51(m,3h),7.65(d,1h,j=8.0hz),8.31(d,1h,j=8.0hz),8.55(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,101.2,103.5,107.9,108.8,117.4,117.7,121.2,122.5,123.6,125.2,125.8,127.7,129.5,129.6,132.1,147.0,148.0,150.6,157.5,164.5;
hrms(esi)calcdfor[m+h]+c21h16no4+:346.10738found346.10468.
实施例十四:
一种4-(4-氟苯基)-5-(4-甲氧基萘-1-基)异噁唑(14)的制备
化合物14的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-氟苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-氟苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-氟苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-氟苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-氟苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物14。
其中化合物14为黄色油状物,收率为58%;且化合物14的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.05(s,3h),6.83-6.91(m,3h),7.15-7.16(m,2h),7.42-7.52(m,3h),7.64(d,1h,j=8.4hz),8.33(d,1h,j=8.0hz),8.60(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.9,103.4,115.8(d,1c,j=21.6hz),117.1(d,1c,j=12.6hz),122.6,125.1,125.8,125.9.126.0,127.8,129.1,129.1,129.2,129.6,129.6,132.0,150.5(d,1c,j=7.0hz),157.6,160.9(d,1c,j=246.2hz),165.0;
hrms(esi)calcdfor[m+h]+c20h15fno2+:320.10813found320.10580.
实施例十五:
一种4-(4-甲氧基-3-硝基苯基)-5-(4-甲氧基萘-1-基)异噁唑(15)的制备
化合物15的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-甲氧基-3-硝基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-甲氧基-3-硝基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-甲氧基-3-硝基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲氧基-3-硝基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-甲氧基-3-硝基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物15。
其中化合物15为黄色固体,收率为86%;且化合物15的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.87(s,3h),4.06(s,3h),6.85-6.87(m,2h),7.23(dd,1h,j=8.8hz,2.4hz),7.42-7.46(m,1h),7.49-7.53(m,2h),7.60(d,1h,j=8.0hz),7.78(d,1h,j=8.0hz),8.33(d,1h,j=8.0hz),8.62(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.9,56.6,103.4,114.0,115.6,116.6,122.5,122.7,124.3,124.8,125.9,126.1,128.0,129.7,131.8,133.0,139.6,150.0,152.2,157.8,165.5;
hrms(esi)calcdfor[m+h]+c21h17n2o5+:377.11320found377.11050.
实施例十六:
一种2-甲氧基-5-(5-(4-甲氧基萘-1-基)异噁唑-4-基)苯胺(16)的制备
化合物16的结构式如下所示:
具体制备步骤:
步骤:将4-(4-甲氧基-3-硝基苯基)-5-(4-甲氧基萘-1-基)异噁唑(化合物15,0.5mmol)、铁粉(1.5mmol)、氯化铵(0.3mmol)、10ml乙醇/水(体积比3:1),回流反应3h,旋干,用硅胶柱层析分离纯化得到化合物16。
其中化合物16为黄色固体,收率为45%;且化合物16的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.77(s,3h),4.04(s,3h),6.56-6.62(m,3h),6.82(d,1h,j=8.0hz),7.41-7.51(m,3h),7.69(d,1h,j=8.0hz),8.31(d,1h,j=8.0hz),8.53(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.5,55.7,103.5,110.7,113.9,117.8,118.0,118.1,122.4,122.7,125.4,125.7,125.9,127.5,129.4,132.4,136.4,146.9,150.6,157.4,164.1;
hrms(esi)calcdfor[m+h]+c21h19n2o3+:347.13902found347.13663.
实施例十七:
一种4-(2-溴苯基)-5-(4-甲氧基萘-1-基)异噁唑(17)的制备
化合物17的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、2-溴苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(2-溴苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(2-溴苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-溴苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(2-溴苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物17。
其中化合物17为黄色油状物,收率为88%;且化合物17的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.98(s,3h),6.72(d,1h,j=8.4hz),6.98(dd,1h,j=8.0hz,2.4hz),7.04-7.11(m,2h),7.36(d,1h,j=8.0hz),7.43-7.49(m,2h),7.60(dd,1h,j=8.0hz,2.4hz),7.85-7.88(m,1h),8.27-8.30(m,1h),8.61(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.7,103.3,116.9,117.1,122.4,123.9,125.2,125.7,125.8,127.6,129.6,131.2,132.0,132.0,133.3,151.9,157.4,166.6;
hrms(esi)calcdfor[m+h]+c20h15brno2+:380.02807found380.02530.
实施例十八:
一种4-(4-溴苯基)-5-(4-甲氧基萘-1-基)异噁唑(18)的制备
化合物18的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、4-溴苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(4-溴苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(4-溴苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-溴苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(4-溴苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物18。
其中化合物18为黄色固体,收率为86%;且化合物18的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.05(s,3h),8.34(d,1h,j=8.0hz),7.06(d,2h,j=8.0hz),7.31(d,2h,j=8.0hz),7.42-7.53(m,3h),7.64(d,1h,j=8.8hz),8.33(d,1h,j=8.4hz),8.60(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,103.4,117.0,117.1,121.6,122.6,125.0,125.8,126.0,127.9,128.8,128.9,129.6,132.0,132.1,150.3,157.7,165.3;
hrms(esi)calcdfor[m+h]+c20h15brno2+:380.02807found380.02505.
实施例十九:
一种4-(3-氯苯基)-5-(4-甲氧基萘-1-基)异噁唑(19)的制备
化合物19的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3-氯苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3-氯苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3-氯苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氯苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氯苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物19。
其中化合物19为黄色油状物,收率为62%;且化合物19的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.05(s,3h),6.83(d,1h,j=8.0hz),7.00-7.02(m,1h),7.09(t,1h,j=8.0hz),7.15-7.18(m,1h),7.26(t,1h,j=2.0hz),7.42-7.53(m,3h),7.64(d,1h,j=8.4hz),8.33(d,1h,j=8.4hz),8.61(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,103.4,116.8,117.0,122.6,125.0,125.6,125.8,126.0,127.3,127.7,127.8,129.7,130.1,131.7,132.0,134.7,150.4,157.7,165.6;
hrms(esi)calcdfor[m+h]+c20h15clno2+:336.07858found336.07626.
实施例二十:
一种4-(3-氟苯基)-5-(4-甲氧基萘-1-基)异噁唑(20)的制备
化合物20的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3-氟苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3-氟苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3-氟苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氟苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-氟苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物20。
其中化合物20为黄色固体,收率为72%;且化合物20的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):4.06(s,3h),6.85(d,1h,j=8.4hz),6.89-6.92(m,2h),6.97(d,1h,j=8.0hz),7.14-7.19(m,1h),7.41-7.46(m,1h),7.48-7.52(m,2h),7.63(d,1h,j=8.4hz),8.33(d,1h,j=8.0hz),8.62(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.9,103.5,114.4,117.0,122.5,123.0,123.2,125.0,125.8,126.0,127.8,129.7,130.5,131.9,132.0,150.3,150.4,157.7,161.7(d,1c,j=244.5hz),165.6;
hrms(esi)calcdfor[m+h]+c20h15fno2+:320.10813found320.10583.
实施例二十一:
一种4-(3-碘-4-甲氧基苯基)-5-(4-甲氧基萘-1-基)异噁唑(21)的制备
化合物21的结构式如下所示:
具体制备步骤:
步骤1:将1-甲氧基萘(1.5mmol)、3-碘-4-甲氧基苯乙酸(1mmol)、三氟乙酸酐(2mmol),加入三氟乙酸5ml,室温搅拌反应12小时,旋干,用硅胶柱层析分离纯化得1-(4-甲氧基萘-1-基)-2-(3-碘-4-甲氧基苯基)乙烷-1-酮。
步骤2:将1-(4-甲氧基萘-1-基)-2-(3-碘-4-甲氧基苯基)乙烷-1-酮(1mmol)、dmf-dma(10mmol)置于反应瓶中,回流反应24小时,旋干,制得(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-碘-4-甲氧基苯基)丙-2-烯-1-酮。
步骤3:将(e)-3-(二甲胺基)-1-(4-甲氧基萘-1-基)-2-(3-碘-4-甲氧基苯基)丙-2-烯-1-酮(1mmol)、盐酸羟胺(1.1mmol)、碳酸钠(1.5mmol)置于反应瓶中,加入5ml甲醇:水(2:1)的混合液做溶剂,再用冰乙酸调节反应液ph为4-5,60~70℃下反应24h,停止反应,倒入饱和碳酸氢钠水溶液中,乙酸乙酯萃取,旋干,用硅胶柱层析分离纯化得到化合物21。
其中化合物21为黄色油状物,收率为57%;且化合物21的核磁氢谱、核磁碳谱与质谱的数据如下所示:
1hnmr(400mhz,cdcl3)δ(ppm):3.78(s,3h),4.05(s,3h),6.55(d,1h,j=8.4hz),6.83(d,1h,j=8.0hz),7.00(dd,1h,j=8.8hz,2.0hz),7.44-7.52(m,3h),7.66(d,1h,j=8.4hz),7.28(d,1h,j=2.0hz),8.32(d,1h,j=8.4hz),8.57(s,1h);
13cnmr(100mhz,cdcl3)δ(ppm):55.8,56.4,86.2,103.4,110.9,116.2,117.2,122.5,124.2,125.2,125.8,126.0,127.8,128.7,129.6,132.0,138.2,150.4,157.5,157.6,164.7;
hrms(esi)calcdfor[m+h]+c21h17ino3+:458.02476found458.02042.
为了进一步验证本发明的优异效果,发明人还进行了如下实验对比:
实验一:
将人类乳腺癌细胞(mcf-7)接种于96孔板中,并在37℃下培养24小时,然后分别加入dmso、顺铂或者不同浓度的目标化合物,继续培养48小时。再将mtt加入到每个孔中,在37℃继续培养4小,小心吸去上清液,并加入dmso溶解沉淀,在经振荡器混匀、溶解后,在酶标仪上检测波长为570nm的吸光度,计算ic50,结果见表1。
表1.萘环-异噁唑型化合物的抑制肿瘤细胞增殖活性(ic50)
由表1可以看出大部分萘环-异噁唑型化合物具有良好的抑制肿瘤细胞增殖的活性,以萘环-异噁唑型化合物1、2、3、7、8、9、10、13、16、18和21活性较好。其中萘环-异噁唑型化合物10的活性最好,对肿瘤细胞mcf-7的抑制ic50为1.23±0.16μm,活性优于阳性对照药物顺铂(15.24±1.27μm)。
实验二:
将纯化后的微管蛋白和不同浓度的萘环-异噁唑型化合物10或秋水仙碱加入到含有1mmgtp和5%甘油的pem缓冲液中(100mmpipes、1mmmgcl2和1mmegta)。将混合物立即置于37℃恒温酶标仪中,于340nm波长处测定吸光度的变化,每分钟测定1次,连续测定20分钟。将吸光度与时间做曲线,观察萘环-异噁唑型化合物10对微管蛋白聚合的影响,处理数据,实验结果如图2所示。
由图2可以看出,实施例十所制备的萘环-异噁唑型化合物10与阳性对照秋水仙碱作用结果相似,具有较好的抑制微管蛋白聚合的活性,随着化合物浓度的提高,对微管蛋白聚合的抑制活性越强,呈现出一种剂量依存关系,是一种新型的微管蛋白抑制剂,抑制ic50为3.4μm。
上述只是本发明的较佳实施例,并非对本发明作任何形式上的限制。任何熟悉本领域的技术人员,在不脱离本发明技术方案范围的情况下,都可利用上述揭示的技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明技术实质对以上实施例所做的任何简单修改、等同变化及修饰,均应落在本发明技术方案保护的范围内。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!