一种使用撞击流反应器制备聚酰亚胺前驱体及其薄膜的制备方法与流程
本发明涉及聚酰亚胺前驱体聚酰胺酸、聚酰亚胺混合薄膜,特别涉及一种用于柔性显示的一种使用撞击流反应器制备聚酰亚胺前驱体的制备方法。
背景技术:
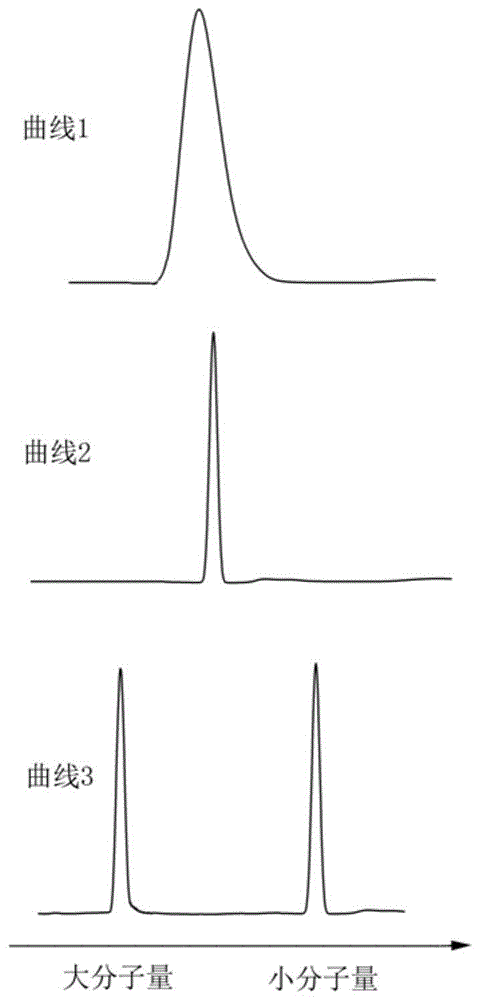
:聚酰亚胺(pi)薄膜具有轻质且柔韧的特性,可作为玻璃基板的替代品应用于面板显示领域。随着柔性显示屏制造技术的不断推进,对于pi薄膜材料也提出了更高的要求,特别是在制作pi层粘合无机层的复合层时,不仅需要pi有更优异的耐热性、尺寸稳定性、耐化学腐蚀性和机械强度,还需要pi层与无机层之间具有更好的粘附力。一般而言,分子量较小的pi树脂和衬底的湿润效果好,在相同的条件下的烘烤过程中通过热布朗运动树脂分子可以和衬底分子达到紧密接触,所以与衬底粘附力强,但是分子量较小的pi树脂内聚力较弱,从而表现为抗腐蚀性较差和机械性能不足。另一方面,分子量较大的pi树脂内聚力较强,从而表现为抗腐蚀性较强和机械性能好,但是分子量较大的pi树脂和衬底的湿润效果差,在相同的条件下的烘烤过程中通过热布朗运动树脂分子不能和衬底分子达到紧密接触,所以与衬底粘附力弱。为改善pi薄膜与无机层之间的粘附力,通常在聚酰胺酸中加入硅烷偶联剂或在聚酰胺酸单体中引入硅氧烷结构。虽然硅烷偶联剂的加入确实能明显的改善pi层与无机层之间的结合力,但偶联剂中通常含有硅元素或者一些脂肪族结构,因此在经过高温固化后,pi薄膜往往含有较高浓度的硅元素,同时由于脂肪族结构的存在,使聚酰胺酸在固化过程中出现较高的挥发物溢出。因此在一些特殊领域的应用中,限制了该方法的使用,例如amoled的制成过程。由于热固化所得聚酰亚胺与很多材料表面的结合力较弱,因此往往需要加入偶联剂来改善聚酰亚胺与一些材料的结合力。但偶联剂的加入往往会导致聚酰亚胺的性能发生变化,从而限制了偶联剂的使用。常规的方法合成聚酰胺酸时,其多分散系数(pdi)一般难以降至很低的值,例如常规方法合成的聚酰胺酸树脂若其中包含了少量的高分子量部分和少量的低分量部分(如图1中曲线1所示),那中间部分就是大量的润湿效果不够和内聚力也不够的组分。因此降低聚酰胺酸树脂的pdi至较低的值(如图1中曲线2所示)对于提升其膜的性能具有重大意义,特别是将pdi小的高分子量聚酰胺酸浆料和pdi小的低分子量聚酰胺酸浆料进行混合后,就可以得到tandem型分子量分布(如图1中曲线3所示)的聚酰胺酸树脂,该树脂浆料固化后得到的膜既具有较好的机械性能也与无机层具有较好的粘附力。技术实现要素:本发明利用撞击流反应器将酸二酐和二胺在撞击流作用下连续反应,成功地制备出一种分子量和分子量分布在较宽范围内可调的聚酰胺酸溶液。该聚酰胺酸溶液的分子量和分子量分布(即多分散系数pdi)可由撞击流速度及酸二酐与二胺的比例来调控,进而可以分别合成pdi小分子量低和pdi小分子量高的聚酰胺酸溶液,组合成tandem型聚酰胺酸溶液。tandem型聚酰胺酸溶液固化后所得到的pi薄膜不仅机械性能优异,还与无机层的粘附力大幅改善。本发明方法中,首先将反应混合液投入到撞击流反应器的预混罐中,进行预混合。然后,通过泵将溶液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度和撞击流速,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应连续的进行,一定反应时间反应完毕后,使反应液从放料口流出,得到分子量可控的聚酰胺酸溶液。本发明利用撞击流反应器来制备极大地强化了微观分子水平上的混合和传质过程,由于反应器内物料高速碰撞,增加了反应物料的接触面积,使传热、传质速率极大增加,利用撞击流反应器在撞击流作用下,通过更加均质的反应环境进行聚合反应制得,所得到的聚酰亚胺前驱体,其分子量分布均匀、多分散系数低。本发明使用撞击流反应器制备聚酰亚胺前驱体聚酰胺酸溶液及利用聚酰胺酸溶液制备复合聚酰亚胺薄膜的方法,所述方法包括:(1)将二胺b加入到有机溶剂e中溶解或者悬浮,然后通过高速分散使得b均匀的分散或溶解于溶剂e中;(2)将酸二酐a加入到有机溶剂e中溶解或者悬浮,然后通过高速分散使得a均匀的分散于溶剂e中;(3)将上述步骤(1)和(2)制备的两种组分混合液及其它添加剂投入到撞击流反应器的预混罐中,进行预混合;然后通过泵将溶液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制撞击流反应器的撞击流速度、反应温度、反应时间,聚合结束后得到分子量可控的聚酰胺酸溶液d;调控反应物之间的配比和撞击流速可得到重均分子量较低的聚酰胺酸溶液和重均分子量较高的聚酰胺酸溶液,重均分子量较低的聚酰胺酸溶液和重均分子量较高的聚酰胺酸溶液混合均匀即得tandem型聚酰胺酸溶液。(4)将上述步骤(3)所得重均分子量较低的聚酰胺酸溶液和重均分子量较高的聚酰胺酸溶液混合均匀后经涂膜、固化即可得到聚酰亚胺薄膜s1,然后在此薄膜上进行化学气相沉积(cvd),即可在s1表面形成二氧化硅层,再在二氧化硅层的表面进行cvd,即可再形成非晶硅层,然后在此非晶硅层上再次涂布聚酰胺酸溶液混合后的浆料,固化后即可得到可进行柔性面板制备的复合聚酰亚胺薄膜。本发明中所采用的酸二酐a单体为任一可以用于聚酰亚胺合成的酸二酐单体,可以优选使用含芳香结构的酸二酐单体。酸二酐单体的优选实例包括:3,3',4,4'-联苯四羧酸二酐(bpda)和均苯四甲酸二酐(pmda)中的一种或两种。本发明中所采用的二胺b单体为任一可以用于聚酰亚胺合成的二胺单体,可以优选使用含芳香结构的二胺单体。二胺单体的优选实例包括:对苯二胺(pda)和2,2'-二三氟甲基-4,4'-二氨基联苯(tfmb)中的一种或两种。本发明反应体系中酸二酐和二胺的配比为0.90-1.1:1。本发明中所采用的溶剂为极性有机溶剂,其中包括所有的室温或加热可以将聚酰胺酸溶解的有机溶剂。有机溶剂的优选实例包括极性非质子溶剂n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-二乙基甲酰胺、n-甲基-2-吡咯烷酮、1,3-二甲基-2-咪唑啉酮、n-甲基己内酰胺、六甲基磷酰三胺、二甲基亚砜;四氢呋喃、丙二醇单甲醚、丙二醇单乙醚、二甘醇单甲醚、二甘醇单乙醚、二甘醇甲基乙基醚、二甘醇二甲醚等米磊溶剂;丙酮、甲基乙基酮、二异丁基酮、二丙酮醇、环己酮等酮类溶剂;乙酸乙酯、丙二醇单甲醚乙酸酯、乳酸乙酯等酯类溶剂,所用溶剂包括以上单一溶剂或几种溶剂的混合溶剂。所制备的聚酰胺酸溶液可以含有酰亚胺催化剂。作为酰亚胺催化剂,可含有吡啶结构的化合物或含有咪唑结构的化合物。其中,吡啶类化合物优选实例包括喹啉、异喹啉、吡啶、吖啶、喹喔啉、6-叔丁基喹啉、3-羟基吡啶、6-喹啉羧酸、3,4-二甲基吡啶。其中,咪唑类化合物优选实施例包括2-苯基咪唑、1-甲基咪唑、1,2-二甲基咪唑、4-乙基-2-甲基咪唑、2-乙基-4甲基咪唑、2-甲基咪唑和1-甲基-4乙基咪唑。以上酰亚胺催化剂可以单独使用,也可以几种混合使用。酰亚胺催化剂的加入量占所有二酐单体和二胺单体总量的0.01~100%,其中优选0.1~10%。为防止聚酰胺酸发生凝胶作用,所制备的聚酰胺酸溶液可以含有磷酸酯类化合物,例如亚磷酸三苯酯或磷酸三苯酯,磷酸酯类化合物的加入量占总固体量的0.01~5%。本发明所述聚酰胺酸溶液的制备方法,其中聚酰胺酸溶液中重均分子量较低的部分范围在50865~54019内,重均分子量较高的部分范围在121690~140861内,在这个范围的聚酰胺酸制备的膜的机械性能和粘附力同时具有适用生产实际的使用价值;在本发明用于合成聚酰胺酸溶液的方法中,需要将酸二酐或二胺单体溶解或分散于溶剂中,形成高速分散的溶液或悬浮液。为了控制悬浮液分散的程度,高速分散的转速优选在3000~10000rpm/min的转速范围内,得到的悬浮液中的颗粒大小在50~300nm,再将高速分散后的溶液或悬浮液进行撞击流反应,该步骤反应的温度在10℃至80℃之间,更优选25℃至60℃之间,步骤(3)中撞击流速控制在200ml/s-700ml/s,反应时间6-50小时。本发明所述的制备方法,所得到的聚酰胺酸分子量分布均匀,多分散性系数在1.100~1.175之间;最终得到的聚酰胺酸溶液粘度在1~7pa·s之间,更优选3~6pa·s之间,固含量在8~45wt%之间,更优选10~20wt%之间,在该固含量和粘度范围内的聚酰胺酸溶液可以更方便的涂膜且涂覆性能优异,从而轻松地制备出膜厚位于5-20μm范围的聚酰亚胺薄膜。本发明所述的制备方法中也涉及到浆料的固化工艺,固化过程中的固化温度在80~600℃范围内,一般优选的固化温度在120~500℃之间,且固化的程序一般分为至少两个阶段,第一阶段为升温到180~280℃之间,然后保温10~60min,第二个阶段升温到400~500℃之间,保持温度10~60min。本发明制备的聚酰亚胺复合薄膜,二氧化硅层和非晶硅层的厚度在第一层pi薄膜与二氧化硅层的粘附力在0.82~1.08n/cm之间,第二层聚酰胺酸薄膜与非晶硅层的粘附力在0.42~0.58n/cm之间。由本发明制备的聚酰亚胺薄膜具有优异的力学性能,可以轻易的制备出拉伸强度达到336mpa以上,拉伸模量达到7.09gpa以上,断裂伸长率达到14.26%以上的聚酰亚胺薄膜。本发明的聚酰胺酸溶液可应用于柔性线路板、柔性amoled基板、太阳能电池板等领域,当选择合适的单体结构时,尤其适用于柔性amoled基板。由本发明所得聚酰胺酸溶液固化所得聚酰亚胺薄膜具有优异的力学特性、优异的尺寸稳定性、与玻璃基板具有良好的粘附性、优异的耐弯折特性和优异的热稳定性,因此十分适用于amoled基板材料。由本专利所述方法得到的聚酰胺酸溶液具有多分散性系数和分子量在很宽的范围内可调,同时又能保证由该聚酰胺酸固化所得聚酰亚胺薄膜具有优异的综合性能。特别是,明显地改善了固化后的聚酰亚胺薄膜与无机层之间的粘附力。附图说明图1是聚酰胺酸浆料分子量分布示意图;其中,曲线1是常规方法合成的聚酰胺酸树脂若其中包含了少量的高分子量部分和少量的低分量部分示意图,曲线2是pdi小的聚酰胺酸分子量分布示意图,曲线3是将pdi小的高分子量聚酰胺酸浆料和pdi小的低分子量聚酰胺酸浆料进行混合后tandem型分子量分布示意图;图2是本发明撞击流反应器的结构流程图;其中:1-预混罐,2-循环泵,3-进料口,4-导流筒,5-物料撞击区,6-循环出口,7-放料口。具体实施方式以下实施例用于说明本发明,但不用来限制本发明的范围。本发明撞击流反应器的结构流程如图2所示。首先分别将已分散好的反应混合液投入到撞击流反应器的预混罐1中,进行预混合。然后,通过循环泵2将混合液输入到撞击流反应器两侧的进料口3,混合液在压力作用下高速经导流筒4流向容器的中心面,并在中心处5发生正面撞击反应。经撞击后的液体物料从两侧的循环出口6流回预混罐,再通过循环泵输送到撞击流反应器进行反应,实现反应连续的进行。反应完毕后,使反应液从放料口7流出。以下为实施例和比较例中使用的化合物的缩写。bpda:3,3',4,4'-联苯四羧酸二酐pmda:均苯四甲酸二酐tfmb:2,2'-二三氟甲基-4,4'-二氨基联苯pda:对苯二胺nmp:n-甲基-2-吡咯烷酮实施例1步骤1:将320.23g(1.0mol)tfmb和1814.64gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到tfmb的分散液或溶液;将270.68g(0.92mol)bpda和1533.85gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到bpda的分散液或溶液。然后将tfmb分散液或者溶液、2.95g的吡啶和bpda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在600ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d1。仅将实施例1中步骤1中吡啶的用量调整为0g,即可得到浆料d2。仅将实施例1中步骤1中吡啶的用量调整为3.06g,bpda的用量调整为291.28g(0.99mol)和bpda分散液或溶液的用量调整为1941.87g,即可得到浆料d3。将100g的浆料d1和500g的浆料d3混合均匀,得到浆料l1。实施例2将300g的浆料d1和300g的浆料d3混合均匀,得到浆料l2。实施例3将500g的浆料d1和100g的浆料d3混合均匀,得到浆料l3。实施例4步骤1:将108.14g(1.0mol)pda和612.79gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到pda的分散液或溶液;将270.68g(0.92mol)bpda和1533.85gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到bpda的分散液或溶液。然后将pda分散液或者溶液、1.89g的吡啶和bpda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在600ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d4。仅将实施例4中步骤1中吡啶的用量调整为2.00g,bpda的用量调整为291.28g(0.99mol)和bpda分散液或溶液的用量调整为1941.87g,即可得到浆料d5。将100g的浆料d4和500g的浆料d5混合均匀,得到浆料l4。实施例5将300g的浆料d4和300g的浆料d5混合均匀,得到浆料l5。实施例6将500g的浆料d4和100g的浆料d5混合均匀,得到浆料l6。实施例7步骤1:将320.23g(1.0mol)tfmb和1814.64gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到tfmb的分散液或溶液;将200.67g(0.92mol)pmda和1137.13gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到pmda的分散液或溶液。然后将tfmb分散液或者溶液、2.60g的吡啶和pmda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在600ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d6。仅将实施例7中步骤1中吡啶的用量调整为2.68g,pmda的用量调整为215.94g(0.99mol)和pmda分散液或溶液的用量调整为1439.59g,即可得到浆料d7。将100g的浆料d6和500g的浆料d7混合均匀,得到浆料l7。实施例8将300g的浆料d6和300g的浆料d7混合均匀,得到浆料l8。实施例9将500g的浆料d6和100g的浆料d7混合均匀,得到浆料l9。实施例10步骤1:将108.14g(1.0mol)pda和612.79gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到pda的分散液或溶液;将200.67g(0.92mol)pmda和1137.13gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到pmda的分散液或溶液。然后将pda分散液或者溶液、1.54g的吡啶和pmda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在600ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d8。仅将实施例10中步骤1中吡啶的用量调整为1.62g,pmda的用量调整为215.94g(0.99mol)和pmda分散液或溶液的用量调整为1439.59g,即可得到浆料d9。将100g的浆料d8和500g的浆料d9混合均匀,得到浆料l10。实施例11将300g的浆料d8和300g的浆料d9混合均匀,得到浆料l11。实施例12将500g的浆料d8和100g的浆料d9混合均匀,得到浆料l12。实施例13步骤1:将320.23g(1.0mol)tfmb和1814.64gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到tfmb的分散液或溶液;将270.68g(0.92mol)bpda和1533.85gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到bpda的分散液或溶液。然后将tfmb分散液或者溶液、2.95g的吡啶和bpda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在450ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d10。仅将实施例13中步骤1中吡啶的用量调整为3.06g,bpda的用量调整为291.28g(0.99mol)和bpda分散液或溶液的用量调整为1941.87g,即可得到浆料d11。将100g的浆料d10和500g的浆料d11混合均匀,得到浆料l13。实施例14步骤1:将320.23g(1.0mol)tfmb和1814.64gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到tmfb的分散液或溶液;将270.68g(0.92mol)bpda和1533.85gnmp在30℃下进行搅拌溶解,然后将此混合物快速加入到高速分散机中进行分散,通过2h的分散,得到bpda的分散液或溶液。然后将tfmb分散液或者溶液、2.95g的吡啶和bpda分散液或者溶液投入到撞击流反应器的预混罐中,进行混合。通过泵将混合液输入到撞击流反应器的进料口,液体流在压力作用下高速经导流筒流向容器的中心面,并在中心处发生正面撞击进行反应,控制反应温度为40℃,系统内的压力为0.3mpa,撞击流速度维持在300ml/s,经撞击后的液体物料从两侧出口流回预混罐,再通过泵输送到撞击流反应器的进料口进行反应,实现反应的连续进行,反应时间为24h,停止反应后,出料得到浆料d12。仅将实施例14中步骤1中吡啶的用量调整为3.06g,bpda的用量调整为291.28g(0.99mol)和bpda分散液或溶液的用量调整为1941.87g,即可得到浆料d13。将100g的浆料d12和500g的浆料d13混合均匀,得到浆料l14。对比例1在干燥的氮气流下,将320.23g(1.0mol)tfmb、2.95g吡啶和1814.64gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入270.68g(0.92mol)bpda和1533.85gnmp,于40℃下反应24小时,停止反应,得到浆料l15。对比例2在干燥的氮气流下,将320.23g(1.0mol)tfmb、3.00g吡啶和1814.64gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入279.51g(0.95mol)bpda和1583.89gnmp,于40℃下反应24小时,停止反应,得到浆料l16。对比例3在干燥的氮气流下,将320.23g(1.0mol)tfmb、3.06g吡啶和1814.64gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入291.28g(0.99mol)bpda和1650.59gnmp,于40℃下反应24小时,停止反应,得到浆料l17。对比例4将100g的浆料l15和500g的浆料l17混合均匀,得到浆料l18。对比例5在干燥的氮气流下,将108.14g(1.0mol)pda、1.89g吡啶和612.79gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入270.68g(0.92mol)bpda和1533.85gnmp,于40℃下反应24小时,停止反应,得到浆料d14。在干燥的氮气流下,将108.14g(1.0mol)pda、2.00g吡啶和612.79gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入291.28g(0.99mol)bpda和1650.59gnmp,于40℃下反应24小时,停止反应,得到浆料d15。将100g的浆料d14和500g的浆料d15混合均匀,得到浆料l19。对比例6在干燥的氮气流下,将320.23g(1.0mol)tfmb、2.60g吡啶和1814.64gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入200.67g(0.92mol)pmda和1137.13gnmp,于40℃下反应24小时,停止反应,得到浆料d16。在干燥的氮气流下,将320.23g(1.0mol)tfmb、2.68g吡啶和1814.64gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入215.94g(0.99mol)pmda和1223.65gnmp,于40℃下反应24小时,停止反应,得到浆料d17。将100g的浆料d16和500g的浆料d17混合均匀,得到浆料l20。对比例7在干燥的氮气流下,将108.14g(1.0mol)pda、1.54g吡啶和612.79gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入200.67g(0.92mol)pmda和1137.13gnmp,于40℃下反应24小时,停止反应,得到浆料d18。在干燥的氮气流下,将108.14g(1.0mol)pda、1.62g吡啶和612.79gnmp加入到反应釜中进行机械搅拌2小时后,向其中加入215.94g(0.99mol)pmda和1223.65gnmp,于40℃下反应24小时,停止反应,得到浆料d19。将100g的浆料d18和500g的浆料d19混合均匀,得到浆料l21。聚酰亚胺薄膜的制备将实施例1~实施例14和对比例1~对比例7所提供的聚酰胺酸浆料分别通过刮刀在玻璃/非晶硅基板上均匀的刮涂成膜,将刮涂所得湿膜在烘箱中从室温以10℃/min升温至200℃,然后在200℃保温50min,再以10℃/min升温至400℃,然后在400℃下保温50min,即可得到固化后聚酰亚胺薄膜,随后进行各项评价。试验例1本试验例提供实施例1~实施例14和对比例1~对比例7所提供的聚酰亚胺薄膜的性能评价。(1)固含量测试样品均匀的涂覆在玻璃容器中,称量样品质量m1。将涂覆好的样品放在烘箱中加热,在100℃下保温30min后,以5℃/min升温至350℃,并在350℃下保温30min。待样品冷却后称量样品重量m2。样品的固含量按照下列公式计算:固含量=(m2/m1)×100%(2)机械性能测试采用万能材料试验机(英斯特朗3360)测得,测试样品宽度10mm,夹具间隔50mm,测试速度为50mm/min,测试样品数为10组。(3)与无机层粘附力测试测试环境为温度25℃,湿度50%±5%,聚酰亚胺浆料在玻璃/非晶硅基板上经高温固化且放置24h后,样品尺寸为宽25mm长200mm,通过拉力机(岛津ags-x,传感器上限为200n)以样品方向180°,按照100mm/min的速率剥离聚酰亚胺薄膜,得到的数据即为聚酰亚胺薄膜与无机层的粘附力。(4)凝胶渗透色谱(gpc)检测采用日本岛津gpc-20a凝胶色谱仪测得。表1是本发明实施例和对比例得到的不同浆料的固含量、数均分子量、重均分子量、多分散系数性能数据统计表。表1浆料的性能浆料固含量数均分子量重均分子量多分散系数d115%46806524691.121d215%45329509961.125d315%1225121358661.109d415%47900536481.120d515%1249091385241.109d615%47238529541.121d715%1241561375651.108d815%48145540191.122d915%1270161408611.109d1015%44709516841.156d1115%1118581284131.148d1215%43289508651.175d1315%1041871216901.168d1415%32646465211.425d1515%872431223151.402d1615%31906456891.432d1715%865071211961.401d1815%32912469981.428d1915%895891256931.403l1515%32231459621.426l1615%53305754261.415l1715%860441206331.402从表1的结果可以明显地看出,通过撞击流反应器制得的聚合物,其pdi较小,这为组合成tandem型聚酰胺酸溶液提供了便捷的方法。此外,撞击流速度越大,得到的聚合物的pdi值越小,分子量越大。表2是利用本发明实施例和对比例得到的不同浆料制备聚酰亚胺混合薄膜的性能数据统计表。表2聚酰亚胺混合薄膜的性能从表2可以得知,常规方法合成的聚酰胺酸浆料通过固化后得到的膜,其与无机层的粘附力和自身的机械性能无法兼顾,而通过本发明撞击流反应器制备的tandem型聚酰胺酸浆料得到的膜,其不仅与无机层具有较好的粘附力而且自身的机械性能非常优异。虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种聚酰亚胺复合材料及其制备方法与制造工艺
- 树脂前体及含有其的树脂组合物、聚酰亚胺树脂膜、树脂薄膜及其制造方法与制造工艺
- 使用了2‑苯基‑4,4’‑二氨基二苯基醚类的清漆、成形性优异的酰亚胺树脂组合物及具有优异断裂伸长率的固化树脂成形体、以及使用了这些的预浸料、酰亚胺预浸料、及耐热性和机械强度优异的纤维强化材料的制造方法与工艺
- 球状氧化铝粉末以及使用该球状氧化铝粉末的树脂组成物的制造方法与工艺
- 用于高TG聚酰亚胺覆铜板的树脂材料配方的制造方法与工艺
- 一种BPADA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种HQDA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种BPADA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种PMDA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种ODPA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺