一种耐高温超长链粘弹性表面活性剂及其制备方法与应用与流程
本发明属于油气开采领域,具体涉及一种粘弹性表面活性剂及制备方法与作为变黏酸液稠化剂的应用。
背景技术:
酸化、酸压技术是恢复和提高低渗油气井产量的有效办法,但长期以来,酸化、酸压施工过程中一直存在如下几个问题:(1)酸岩反应速度过快,导致岩层深部酸化难以实现;(2)在基质酸化过程中,由于储层纵向渗透率差异大,酸液优先流入高渗层段,导致高渗层段过度溶蚀,而需要改造的低渗地层段几乎得不到处理;(3)在压裂酸化(酸压)过程中酸液滤失严重,且难以控制。石油工业界研究的变粘酸液体系为解决上述问题提供了新的思路。变粘酸液是由酸液、稠化剂、酸液添加剂(粘土稳定剂、缓蚀剂、铁离子稳定剂)等三部分组成,在酸化、酸压过程中酸液粘度随ph变化,能较好地平衡酸液流动,使不同渗透率的地层均能达到酸化目的。根据稠化剂的类型,变粘酸液可分为两种类型:基于聚合物稠化剂的酸液和基于粘弹性表面活性剂(viscoelasticsurfactant,ves)稠化剂的酸液(ves类变粘酸)。其中基于ves的变粘酸液的作用机理是:酸液泵入地层首先进入高渗地层并与岩石发生反应,随着酸液的消耗,ph值升高,酸、岩反应产生金属离子(ca2+、mg2+),酸液中表面活性剂的微观聚集体结构发生改变,从球状胶束转变为蠕虫状胶束,蠕虫状胶束继而相互缠绕形成三维空间网状结构,使酸液体系黏度剧增,实现变粘过程。高黏度酸液体系封堵高渗层段后,后续酸液自动转向低渗层段,从而实现对酸液的分流转向;对目的层处理结束后,高黏度酸液遇到储层中的碳氢化合物自动破胶。与聚合物类变粘酸相比,ves类变粘酸液具有易配置、易破胶、对地层伤害低等优点,且酸液体系中不含有金属阳离子交联剂,避免了酸液消耗后ph值升高而产生金属氢氧化物沉淀及含硫井中产生金属硫化物沉淀的问题。而对于ves类变粘酸液,ves类稠化剂的类型及性能对变粘酸液的施工效果具有非常重要的影响。
目前常用的ves类稠化剂主要是两性表面活性剂(如甜菜碱和氧化叔胺两类表面活性剂)或阳离子季铵盐类表面活性剂(如长链烷基季铵盐和长链烷基卤化吡啶)。随着目前油气开发技术和开采需求的不断提高,井深不断增加,温度越来越高,现有ves类稠化剂的耐温性难以满足应用要求,在120℃左右酸液粘度迅速下降。并且现有ves类稠化剂在强酸环境中化学结构不稳定,含有易水解等弱键(酰胺键、酯键)的ves稠化剂难易长时间保持稳定的化学结构,从而失去增粘效果。
赵梦云等(赵梦云,赵忠扬,赵青.变粘酸用转向稠化剂vca的研制.油田化学,2005,22(2):133-136)制备了一种基于长链脂肪酸衍生物的阳离子表面活性剂,以其为稠化剂配制的变粘酸液虽初始粘度很低,但当酸液体系内盐酸与碳酸盐岩反应后,酸液中ca2+浓度增加,导致体系的粘弹性急剧增加,但在90℃、170s–1下恒温剪切1h后,粘度仅为20mpa·s,说明该稠化剂耐温性不好。董景锋等人(董景锋,阿不都卡德尔·阿不都热西提,李晓艳,古丽加纳提·阿扎提.一种新型两性表面活性剂自转向酸体系.钻井液与完井液,2016,(01):102-106)以两性表面活性剂为稠化剂配制稠化剂浓度为5%的ves变粘酸液在25~85℃的温度区间内,酸液黏度基本保持稳定,但当温度高于90℃时,酸液粘度随着温度的升高即快速下降。
此外,现有变粘酸液中ves稠化剂的质量分数需要达到5%以上才能保证酸液有较高粘度,经济成本较高。
技术实现要素:
本发明的目的在于针对现有技术的不足,提供一种耐高温粘弹性超长链表面活性剂及其制备方法与应用,该粘弹性超长链表面活性剂作为变黏酸液稠化剂具有优异的酸液增稠能力和良好的耐温性,且化学结构稳定,满足强酸、高温等恶劣环境的应用。
本发明所述粘弹性超长链表面活性剂,其结构式如下:
结构式中,r1为具有18~22个碳原子的饱和或不饱和烷烃,r2为h原子,或具有2~4个碳原子的饱和烷烃,r3为h原子或具有2~4个碳原子的饱和烷烃,r2、r3可相同或不同,x—为cl-、br-、f-、co32-、so42-、hcoo-和ch3coo-中的一种。
本发明提供的上述耐高温粘弹性超长链表面活性剂的制备方法,步骤如下:
(1)将脂肪酸溶于二氯甲烷中,再加入有机胺、羰基活化剂,待完全溶解后加入催化剂4-二甲氨基吡啶于室温下搅拌反应24~36h,反应结束加入二氯甲烷稀释反应液,再依次用质量浓度为0.1%~0.5%的hcl溶液、饱和nahco3溶液、饱和nacl溶液分别洗涤,然后收集有机相,将有机相经无水mgso4干燥后过滤,将滤液除去溶剂得到脂肪酰胺;
其中,反应前二氯甲烷的用量应使脂肪酸、有机胺、羰基活化试剂和催化剂完全溶解;有机胺与脂肪酸的摩尔比为(1.1~1.5):1;催化剂4-二甲氨基吡啶与脂肪酸的摩尔比为(0.2~0.5):1;羰基活化剂与脂肪酸的摩尔比为(2~2.5):1;
(2)将所得脂肪酰胺溶于四氢呋喃中,在0~5℃下滴入金属氢化物的四氢呋喃溶液形成反应液,金属氢化物的四氢呋喃溶液的滴入量应使反应液中脂肪酰胺与金属氢化物的摩尔比为1:(3.0~3.5),滴加完毕后升温至70~150℃反应24~36h,反应结束后向所得反应液中依次加入去离子水、质量浓度10%~20%的naoh溶液、去离子水将反应淬灭,然后过滤,所得滤液用无水mgso4干燥后再次过滤,将再次过滤所得滤液除去溶剂得到脂肪叔胺;
(3)将脂肪叔胺与质量浓度10%~20%的酸液混合并搅拌均匀,使脂肪叔胺质子化,即形成超长链粘弹性表面活性剂稠化剂,所述酸液的用量为脂肪叔胺与酸液中氢离子的摩尔比为1:1。
上述方法中,所述脂肪酸为具有18~22个碳原子的饱和或不饱和烷基脂肪酸中的至少一种。
上述方法中,所述有机胺为具有4~8个碳原子的饱和烷基有机胺中的至少一种。
上述方法中,所述羰基活化剂为1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc·hcl)、二环己基碳二亚胺(dcc)、n,n-二异丙基碳二亚胺(dic)中的至少一种。
上述方法中,所述金属氢化物为lialh4或nabh4。
上述方法中,所述酸液为hcl溶液、hbr溶液、hf溶液、h2so4溶液、h2co3溶液、hcooh溶液、ch3cooh溶液中的至少一种。
上述方法中,步骤(3)中,将脂肪叔胺与质量浓度10%~20%的酸液混合并搅拌均匀优选在50~80℃下进行。
上述方法中,步骤(1)中加入二氯甲烷稀释反应液时二氯甲烷的用量一般为反应液体积的2~5倍,步骤(2)中四氢呋喃作为溶剂的用量至少能使相应的溶质脂肪酰胺和金属氢化物完全溶解。
本发明所述超长链粘弹性表面活性剂作为稠化剂应用于变黏酸液时,与质量浓度为10%~20%的酸液混合即可得到基于该超长链粘弹性表面活性剂稠化剂的变黏酸液,所述质量浓度为10%~20%的酸液为hcl溶液、hbr溶液、hf溶液、h2so4溶液、h2co3溶液、hcooh溶液、ch3cooh溶液中的至少一种,酸液用量为使得到的变黏酸液中粘弹性超长链表面活性剂的质量浓度为3%~5%。
与现有技术相比,本发明具有以下有益效果:
1、本发明所述超长链粘弹性表面活性剂作为稠化剂不仅具有优异的酸液增稠性能,而且耐温性较好,在温度高达150℃时,基于该稠化剂的变黏酸液体系的黏度较为稳定且大于20mpa·s,满足强酸、高温等恶劣环境的应用需求。
2、本发明所述粘弹性超长链表面活性剂作为稠化剂化学结构稳定性良好(见实施例3),即使长期处于高温强酸性等恶劣环境下也不会发生降解,满足强酸、高温等恶劣环境的应用需求。
3.本发明所述超长链粘弹性表面活性剂作为稠化剂应用于变黏酸液时,添加质量浓度为3%即可达到较好的增粘效果,因此经济性高,利于生产中使用。
4、本发明所述粘弹性超长链表面活性剂制备方法可通过改变脂肪酸、脂肪胺和酸液的种类等,实现对粘弹性超长链表面活性剂稠化剂性能的调节。
附图说明
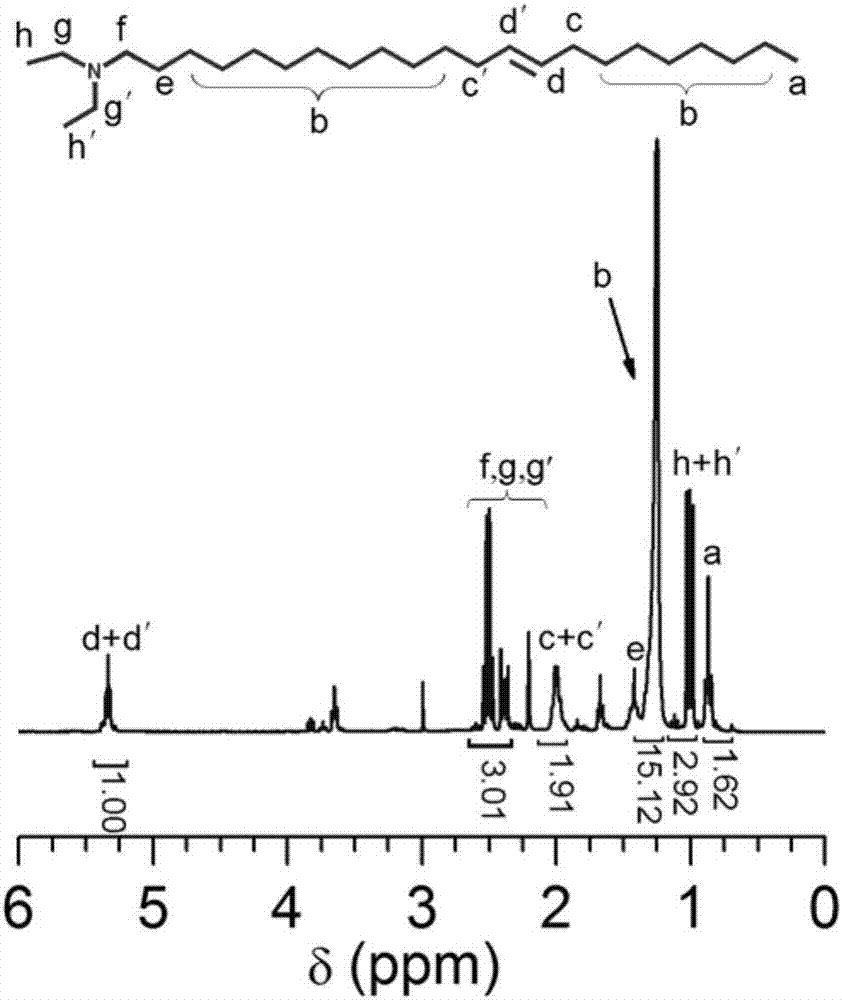
图1是n-(顺-二十二碳-9-烯基)-n,n-二乙基酰胺的1hnmr谱图。
图2是n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺的1hnmr谱图。
图3是3.0%n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐在20%hcl溶液中的流变行为。
图4是3.5%n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐在20%hcl溶液中的流变行为。
图5是流变测试后n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐的1hnmr谱图。
图6是n,n-二乙基-n-正二十二烷基酰胺的1hnmr谱图。
图7是n,n-二乙基-n-正二十二烷基叔胺的1hnmr谱图。
图8是3.0%n,n-二乙基-n-正二十二烷基叔胺盐酸盐在20%hcl溶液中的流变行为图。
图9是流变测试后n,n-二乙基-n-正二十二烷基叔胺盐酸盐的1hnmr谱图。
图10是n,n-二乙基-n-正十八烷基酰胺的1hnmr谱图。
图11是n,n-二乙基-n-正十八烷基叔胺的1hnmr谱图。
图12是3.0%n,n-二乙基-n-正十八叔胺盐酸盐在20%hcl溶液中的流变行为图。
图13是流变测试后n,n-二乙基-n-正十八烷基叔胺盐酸盐的1hnmr谱图。
具体实施方式
下面通过具体实施方式对本发明所述耐高温超长链粘弹性表面活性剂及其制备方法与应用作进一步说明。
以下实施例中的,浓度及含量百分比均为质量百分比。
实施例1
(1)称取16.92g(0.05mol)顺二十二碳-13-烯酸(俗名“芥酸”)溶于二氯甲烷,依次加入4.02g(0.055mol)二乙基胺、19.1g(0.1mol)edc·hcl,待溶解完全后再加入1.22g(0.01mol)dmap,于室温下反应24h。反应结束后,用250ml二氯甲烷稀释反应液,再依次用0.1%的hcl溶液、饱和nahco3溶液和饱和nacl溶液分别洗涤三次,收集有机相,加入无水mgso4干燥2h,过滤,将滤液旋转蒸发除去溶剂,真空干燥,即得到n,n-二乙基-n-正二十二烷基酰胺,其结构表征见1hnmr谱图(图1),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(2)将所得n,n-二乙基-n-正二十二烷基酰胺加入到500ml圆底烧瓶中,加入100ml四氢呋喃溶解,待完全溶解后降温至2℃,逐滴加入lialh4的四氢呋喃溶液,其中脂肪酰胺与金属氢化物的摩尔比为1:3.0,滴加完毕后升温至75℃反应24h,反应结束后向所得反应液中依次加入去离子水、10%naoh溶液、去离子水将反应淬灭,然后过滤反应液,滤液用无水mgso4干燥,过滤,将滤液旋转蒸发除去溶剂,得到n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺,其结构表征见1hnmr谱图(图2),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(3)将n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺与质量浓度15%hcl溶液混合,hcl溶液的用量为使n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺与hcl溶液中氢离子的摩尔比为1:1,50℃搅拌24h至均匀,得到n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐,即为粘弹性超长链表面活性剂。
(4)将n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐与质量浓度15%hcl溶液混合,即获得变粘酸液,hcl溶液的用量为使n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐在变黏酸液中的质量浓度为3%。通过哈克转矩流变仪及其所配套的pz39转子/转筒系统进行测试,在剪切速率为100s-1下测量所得变黏酸液黏度随温度、时间的变化关系,结果见图3。从图3可以看出,当温度稳定在150℃时,溶液表现的黏度较为稳定且大于20mpa·s,表明本实施例所得粘弹性超长链表面活性剂作为稠化剂具有良好的耐温性。
测试结束后,将酸液冷却至室温,旋蒸,冷冻干燥后得到n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐,其结构表征见1hnmr谱图(图5),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物在高温强酸环境下并未发生降解、变质,说明其化学结构稳定性较好,满足高温、强酸性等恶劣环境下的应用要求。
实施例2
本实施与实施例1的不同之处在于,步骤(4)中,hcl溶液的用量为使n-(顺-二十二碳-9-烯基)-n,n-二乙基叔胺盐酸盐在变黏酸液中的质量浓度为3.5%。
在剪切速率为100s-1下测量所得变黏酸液黏度随温度、时间的变化关系,结果见图4。从图4可以看出,当温度稳定在150℃时,溶液表现的黏度较为稳定且大于20mpa·s,表明本实施例所得粘弹性超长链表面活性剂作为稠化剂具有良好的耐温性。
实施例3
(1)称取17.03g(0.05mol)正二十二酸(俗名“花生酸”)溶于二氯甲烷,依次加入4.75g(0.065mol)二乙基胺、21.01g(0.11mol)edc·hcl,待溶解完全后再加入1.83g(0.015mol)dmap,室温下反应30h。反应结束后,用250ml二氯甲烷稀释反应液,再依次用0.2%的hcl溶液、饱和nahco3溶液和饱和nacl溶液分别洗涤三次,收集有机相,加入无水mgso4干燥,过滤,将滤液旋转蒸发除去溶剂,真空干燥,得到n,n-二乙基-n-正二十二烷基酰胺,其结构表征见1hnmr谱图(图5),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(2)将所得n,n-二乙基-n-正二十二烷基酰胺加入到500ml圆底烧瓶中,加入100ml四氢呋喃溶解,待完全溶解后降温至0~5℃,随后逐滴加入lialh4的四氢呋喃溶液,其中脂肪酰胺与金属氢化物的摩尔比为1:3.3,滴加完毕后升温至75℃反应30h。反应结束后逐步加入去离子水、15%naoh水溶液、去离子水将反应淬灭。然后过滤反应液,滤液用无水mgso4干燥、过滤、旋转蒸发除去溶剂得到n,n-二乙基二十二烷基叔胺。其结构表征见1hnmr谱图(图6),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(3)将n,n-二乙基二十二烷基叔胺与质量浓度为15%hcl溶液混合,hcl溶液的用量为使n,n-二乙基二十二烷基叔胺与hcl溶液中氢离子的摩尔比为1:1,50℃搅拌24h至均匀,得n,n-二乙基二十二烷基叔胺盐酸盐,即为粘弹性超长链表面活性剂。
(4)将n,n-二乙基二十二烷基叔胺盐酸盐与质量浓度15%hcl溶液混合,即获得变粘酸液,hcl溶液的用量为使n,n-二乙基二十二烷基叔胺盐酸盐在变黏酸液中的质量浓度为3%。通过哈克转矩流变仪及其所配套的pz39转子/转筒系统进行测试,在剪切速率为100s-1测试所得变黏酸液黏度随温度、时间的变化关系,结果见图7。从图7可以看出,当温度稳定在150℃时,溶液表现的黏度较为稳定且大于30mpa·s,表明本实施例所得表面活性剂具有较好的耐温性。
测试结束后,将酸液冷却至室温,有白色固体析出,过滤,冷冻干燥后得到n,n-二乙基二十二烷基叔胺盐酸盐,其结构表征见1hnmr谱图(图8)。通过分析谱图8,可知n,n-二乙基二十二烷基叔胺盐酸盐在高温强酸环境下并未发生降解、变质,说明其化学结构稳定性较好,满足高温、强酸性等恶劣环境下的应用要求。
实施例4
本实施与实施例3的不同之处在于,步骤(3)和步骤(4)中酸液为质量浓度为10%的乙酸溶液,步骤(4)中乙酸溶液的用量为使n,n-二乙基二十二烷基叔胺乙酸盐在变黏酸液中的质量浓度为4%。
实施例5
(1)称取14.24g(0.05mol)正十八酸(俗名“硬脂酸”)溶于二氯甲烷,依次加入5.48g(0.075mol)二乙基胺、23.87g(0.125mol)edc·hcl,待溶解完全后再加入3.02g(0.025mol)dmap,室温下反应36h。反应结束后,用250ml二氯甲烷稀释反应液,再依次用0.5%的hcl溶液、饱和nahco3溶液和饱和nacl溶液分别洗涤三次,收集有机相,加入无水mgso4干燥,过滤,将滤液旋转蒸发除去溶剂,真空干燥,即得到n,n-二乙基-n-正十八烷基酰胺,其结构表征见1hnmr谱图(图8),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(2)将所得n,n-二乙基-n-正十八烷基酰胺加入到500ml圆底烧瓶中,加入100ml四氢呋喃溶解,待完全溶解后降温至0~5℃,逐滴加入lialh4的四氢呋喃溶液,其中脂肪酰胺与金属氢化物的摩尔比为1:3.5,滴加完毕后升温至75℃反应36h。反应结束后逐步加入去离子水、20%naoh水溶液、去离子水将反应淬灭。然后过滤反应液,滤液用无水mgso4干燥,过滤,将滤液旋转蒸发除去溶剂得到n,n-二乙基正十八叔胺,其结构表征见1hnmr谱图(图9),化合物中各质子峰的化学位移已在谱图中找到归属,且各质子共振峰积分面积比值与理论值之间吻合较好,表明该化合物已成功合成。
(3)将n,n-二乙基正十八叔胺与质量的浓度为15%hcl溶液混合,hcl溶液的用量为使n,n-二乙基正十八叔胺与hcl溶液中氢离子的摩尔比为1:1,50℃搅拌24h至均匀,得到n,n-二乙基正十八叔胺盐酸盐,即为粘弹性超长链表面活性剂。
(4)将n,n-二乙基正十八叔胺盐酸盐与质量浓度15%hcl溶液混合均匀,即获得变粘酸液,hcl溶液的用量为使n,n-二乙基正十八叔胺盐酸盐在变黏酸液中的质量浓度为3%。通过哈克转矩流变仪及其所配套的pz39转子/转筒系统进行测试,在剪切速率为100s-1测量所得变黏酸液的黏度随温度、时间的变化关系,结果见图11。从图11可以看出,当温度稳定在150℃时,溶液表现的黏度较为稳定且大于20mpa·s。
测试结束后,将酸液冷却至室温,有白色固体析出,过滤,冷冻干燥后得到n,n-二乙基正十八叔胺盐酸盐,其结构表征见1hnmr谱图(图12)。通过分析谱图可知,n,n-二乙基正十八叔胺盐酸盐在高温强酸环境下并未发生降解、变质,表明其化学结构稳定性较好,满足高温、强酸性等恶劣环境下的应用要求。
实施例6
本实施与实施例5的不同之处在于,步骤(3)和步骤(4)中酸液为质量浓度为20%的硫酸溶液,步骤(4)中硫酸溶液的用量为使n,n-二乙基正十八叔胺盐在变黏酸液中的质量浓度为5%。
- 还没有人留言评论。精彩留言会获得点赞!