耐硫甲烷化催化剂及其制备方法和甲烷化方法与流程
本发明涉及耐硫甲烷化催化剂领域,具体涉及一种含有单斜相氧化锆的低温耐硫甲烷化催化剂,制备该催化剂的燃烧法,和该催化剂在低温含硫条件下进行甲烷化的方法。
背景技术:
天然气具有高热值、低碳排放、易长途输送等特点,世界上多数发达国家均将其列为首选燃料。我国“富煤,缺油,少气”的资源结构导致天然气供求矛盾突出。而我国煤炭资源相对丰富,将煤转化为天然气不但可以实现煤资源洁净转化,还能有效补充国内的天然气供应。
现有的甲烷化技术通常采用间接甲烷化工艺和ni基催化剂。但ni基催化剂易积碳且对硫毒物非常敏感,为了延缓催化剂因积碳和硫中毒导致的失活,在原料气进入甲烷化装置前需预先进行水煤气变换、酸性气体分离以及精脱硫等工艺。在以mo基催化剂为核心的直接甲烷化工艺中,催化剂在含硫气氛下活性相为mos2,由于mo基催化剂兼具甲烷化和水汽变换活性,并且具有优越的抗积碳性能,因此通过煤气化得到的含硫合成气则无需进行水煤气变换和酸性气体脱除,直接进行酸性甲烷化反应,这极大地简化了工艺流程。
甲烷化催化剂是甲烷化技术的核心。但是相比ni基催化剂,mo基催化剂的活性相对较低,这是限制其工业应用的主要瓶颈所在。
合成气甲烷化反应是强放热反应,受热力学平衡影响,在第1-2段反应器中进行高温反应时合成气转化不能进行完全,必须在后续工艺增加1-2段反应器进行中低温反应,于相对较低温度下将未转化的合成气转化完全。在耐硫直接甲烷化反应中,co与h2通过反应(2co+2h2—→ch4+co2)合成ch4,在多段甲烷化工艺最后1-2段中,由于系统中作为产物的co2逐渐累积,易导致逆水汽变换(h2+co2—→co+h2o)等副反应,从而影响ch4的生成,因此催化剂不仅要求具有较高的甲烷化催化活性,同时还需具有低逆水汽变换活性,即能够对逆水汽变换反应不敏感。
cn103433026a公开了一种zro2负载的高温定性耐硫甲烷化催化剂,包括5-25份(重量)moo3,3-35份(重量)y2o3,40-92份(重量)zro2。该催化剂可用于多段甲烷化工艺的最后1-2段。该催化剂的制备方法包括:(1)通过沉淀法、沉积沉淀法、或溶胶凝胶法制备zro2载体或选用商购的zro2载体;(2)通过浸渍法或沉积沉淀法将催化剂助剂y2o3的前体溶液负载在上述zro2载体上;(3)在上述催化剂助剂y2o3的前体分解温度下或之上焙烧干燥和浸渍或沉积后的zro2载体,得到负载有催化剂助剂y2o3的zro2载体,其中,浸渍、干燥和焙烧步骤任选地重复多次;(4)通过浸渍法或沉积沉淀法将催化剂活性组分moo3的前体溶液负载在上述负载有催化剂助剂y2o3的zro2载体上;(5)在上述催化剂活性组分moo3的前体分解温度下或之上焙烧干燥和浸渍或沉积后的zro2载体,得到上述负载有催化剂活性组分moo3和催化剂助剂y2o3的高稳定性耐硫甲烷化催化剂,其中浸渍、干燥和焙烧步骤任选地重复多次。该催化剂的性能很大程度取决于载体的纯度和比表面积,但目前具有高比表面积的纯单斜相zro2的制备方法相对较复杂,因而限制了该催化剂的工业应用。
cn105879854a公开了一种使用水热法制备的耐硫甲烷化催化剂,该催化剂包括:mo、w和v中的一种或多种作活性组分,la、ce和y中的一种或多种作载体改性剂,al2o3、sio2或zro2作载体。该催化剂通过将活性组分前驱体、载体改性剂前驱体、载体前驱体与沉淀缓释剂混合后进行水热处理而得,但该方法可能会存在氧化锆载体晶相不纯而导致催化剂性能不稳定,另外制备工艺对设备材质的耐酸性能要求高,同时水耗和能耗较大。
本发明的目的就是提供一种同时兼具高甲烷化活性与稳定性以及低逆水汽变换活性的甲烷化催化剂及其制备方法。
技术实现要素:
本发明的目的是为了克服现有技术的耐硫甲烷化催化剂不能同时兼具高甲烷化活性与稳定性以及低逆水汽变换活性的问题,提供耐硫甲烷化催化剂及其制备方法和甲烷化方法,该耐硫甲烷化催化剂的组成中,氧化锆为单斜晶相,并添加活性助剂和载体改进剂,可以提供催化剂具备耐硫性能下兼具高甲烷化活性与稳定性以及低逆水汽变换活性。
为了实现上述目的,本发明第一方面提供一种耐硫甲烷化催化剂,以该催化剂的总重量为基准,该催化剂含有10~30重量%的氧化钼,1~5重量%的活性助剂,2~10重量%的载体改进剂和55~87重量%的氧化锆;所述活性助剂为选自re、co和ni中的至少一种金属的氧化物,所述载体改进剂选自la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属的氧化物;其中,所述催化剂中,氧化锆仅为单斜晶相。
优选地,所述催化剂的xrd谱图中,2θ为24.2°、28.1°、31.4°、34.2°、50.2°和59.9°处出现单斜相氧化锆的衍射峰。
优选地,氧化钼以mo计,所述活性助剂以金属m1计,氧化锆以zr计,所述载体改进剂以金属m2计,mo:m1:zr:m2的摩尔比为1:(0.05~0.3):(1~7):(0.1~0.5),优选为1:(0.07~0.2):(3~5):(0.2~0.3)。
本发明第二方面提供一种制备本发明的耐硫甲烷化催化剂的方法,包括:
(1)将氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体、活性助剂前驱体、燃烧剂与稳定剂放入去离子水中溶解为混合溶液;
(2)将所述混合溶液进行浓缩至粘稠状,得到透明胶状物;
(3)将所述透明胶状物放置于350~650℃下,在所述燃烧剂的作用下进行燃烧反应0.5~3h;
其中,所述氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体和活性助剂前驱体的用量满足得到的甲烷化催化剂中,含有10~30重量%的氧化钼,1~5重量%的活性助剂,2~10重量%的载体改进剂和55~87重量%的氧化锆;所述活性助剂为选自re、co和ni中的至少一种金属的氧化物,所述载体改进剂选自la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属的氧化物;氧化锆为单斜晶相。
优选地,所述氧化钼前驱体以mo计,所述氧化锆前驱体以zr计,所述活性助剂前驱体以m1计,所述载体改性剂前驱体以m2计,满足mo:m1:zr:m2的摩尔比为1:(0.05~0.3):(1~7):(0.1~0.5),优选为1:(0.07~0.2):(3~5):(0.2~0.3)。
优选地,所述燃烧剂与所述混合溶液中金属总量的摩尔比为(0.7~3):1,优选为(1~2):1。
优选地,所述燃烧剂为尿素、甘氨酸、乙二醇、氨基乙酸、甘油和甘露醇中的至少一种。
优选地,所述稳定剂与所述混合溶液中金属总量的摩尔比为(0.01~0.2):1。
优选地,所述稳定剂为柠檬酸、聚乙烯醇、二乙醇胺和乙酰丙酮中的至少一种。
本发明第三方面,提供一种甲烷化方法,该方法包括:
(a)将本发明的耐硫甲烷化催化剂在含硫还原气氛下,在温度350~450℃、表压压力0.1~0.2mpa下进行预硫化反应2~6h,相对于1g的所述耐硫甲烷化催化剂,所述含硫还原气氛的流量为3~6l/h,所述含硫还原气氛含有硫化氢与氢气,所述含硫还原气氛中硫化氢含量为2~5体积%;
(b)在经步骤(a)得到的预硫化催化剂存在下,将含有氢气、一氧化碳、二氧化碳和硫化氢的混合气进行甲烷化反应,甲烷化反应温度为300~650℃,优选为400~600℃;甲烷化反应压力为0.5~6mpa;所述混合气中,氢气:二氧化碳:一氧化碳的体积比为(0.7~4):(0.5~2):1,硫化氢含量为0.4~0.8体积%,所述混合气的进气体积空速为5000~20000h-1。
通过上述技术方案,本发明提供了一种能在低温、高co2条件下具有高甲烷化活性与稳定性的耐硫甲烷化催化剂。
本发明的优点在于:(1)催化剂制备过程中借助燃烧剂通过燃烧反应一步合成,使催化剂各组分前驱体直接进行高温处理,有利于促进各组分间的相互作用,特别是在组成中添加的活性助剂的协同作用下,有利于增强活性组分钼与载体间的相互作用,从而提高催化剂的反应性能;(2)燃烧过程使用短暂高温,可以有效避免传统焙烧引起的催化剂结构破坏和烧结;(3)制备过程高温燃烧时间较短,同时制备过程无需对催化剂进行多次洗涤,具有制备工艺简单、能耗与水耗低的特点;(4)该方法制备的耐硫甲烷化催化剂中,氧化锆以单斜晶相存在。
附图说明
图1是对比例1-3和实施例1-2所制备催化剂的xrd谱图
图2是实施例1所制备催化剂的sem图片
图3是实施例2所制备催化剂的sem图片
图4是实施例9所制备催化剂的sem图片
图5是实施例1所制备催化剂经硫化后的tem图片
图6是实施例2所制备催化剂经硫化后的tem图片
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明第一方面提供一种耐硫甲烷化催化剂,以该催化剂的总重量为基准,该催化剂含有10~30重量%的氧化钼,1~5重量%的活性助剂,2~10重量%的载体改进剂和55~87重量%的氧化锆;所述活性助剂为选自re、co和ni中的至少一种金属的氧化物,所述载体改进剂选自la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属的氧化物;其中,所述催化剂中,氧化锆仅为单斜晶相。
本发明中,组成所述催化剂的各组分的含量均是以各组分的金属氧化物形式进行计量。优选地,催化剂含有18~24重量%的氧化钼,2~3重量%的活性助剂,4~6重量%的载体改进剂和67~73重量%的氧化锆。
优选地,所述催化剂的xrd谱图中,2θ为24.2°、28.1°、31.4°、34.2°、50.2°和59.9°处出现单斜相氧化锆的衍射峰。
本发明的耐硫甲烷化催化剂中,载体中的氧化锆为单斜晶相的氧化锆,有助于催化剂获得较好的稳定性。
进一步地,所述耐硫甲烷化催化剂中加入载体改性剂,一方面可以提高载体稳定性,抑制载体的相变与烧结,另一方面可以促进活性组分在载体表面的分散,有利于提高催化剂的甲烷化活性与稳定性。
再有所述耐硫甲烷化催化剂的组成中加入活性助剂成分,可以改善钼的分散性能和还原性能,由此可以进一步提高催化剂的甲烷化性能。
本发明中,所述耐硫甲烷化催化剂的组成中上述各组分之间存在一定的用量关系,可以进一步提高催化剂在较高二氧化碳含量和低温下的耐硫甲烷化性能。优选地,氧化钼以mo计,所述活性助剂以金属m1计,氧化锆以zr计,所述载体改进剂以金属m2计,mo:m1:zr:m2的摩尔比为1:(0.05~0.3):(1~7):(0.1~0.5),优选为1:(0.07~0.2):(3~5):(0.2~0.3)。
根据本发明,优选地,所述耐硫甲烷化催化剂的比表面积不小于150m2/g,优选不小于165m2/g;更优选地,所述耐硫甲烷化催化剂的比表面积可以为165~200m2/g,有助于所述耐硫甲烷化催化剂获得较好的甲烷化活性与稳定性。
本发明第二方面提供一种制备本发明的耐硫甲烷化催化剂的方法,包括:
(1)将氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体、活性助剂前驱体、燃烧剂与稳定剂放入去离子水中溶解为混合溶液;
(2)将所述混合溶液进行浓缩至粘稠状,得到透明胶状物;
(3)将所述透明胶状物放置于350~650℃下,在所述燃烧剂的作用下进行燃烧反应0.5-3h;
其中,所述氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体和活性助剂前驱体的用量满足得到的甲烷化催化剂中,含有10~30重量%的氧化钼,1~5重量%的活性助剂,2~10重量%的载体改进剂和55~87重量%的氧化锆;所述活性助剂为选自re、co和ni中的至少一种金属的氧化物,所述载体改进剂选自la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属的氧化物;氧化锆为单斜晶相。
优选地,含有18~24重量%的氧化钼,2~3重量%的活性助剂,4~6重量%的载体改进剂和67~73重量%的氧化锆。
本发明提供的甲烷化催化剂的制备方法中,加入燃烧剂和稳定剂,进行步骤(3)时,可以通过燃烧剂的自发燃烧,影响所述透明胶状物形成最终的耐硫甲烷化催化剂的性能,可以提供得到的耐硫甲烷化催化剂兼具高甲烷化活性与稳定性以及低逆水汽变换活性。优选步骤(3)中,将所述透明胶状物放置于400~500℃。
本发明的制备方法中,优选地,所述氧化钼前驱体以mo计,所述氧化锆前驱体以zr计,所述活性助剂前驱体以m1计,所述载体改性剂前驱体以m2计,满足mo:m1:zr:m2的摩尔比为1:(0.05~0.3):(1~7):(0.1~0.5),优选为1:(0.07~0.2):(3~5):(0.2~0.3)。可以有助于进一步改善得到的耐硫甲烷化催化剂的甲烷化性能。
根据本发明,优选情况下,其中,所述氧化钼前驱体选自硝酸钼、草酸钼、甲酸钼、乙酸钼或钼酸铵。
根据本发明,优选地,所述氧化锆前驱体选自氧氯化锆、硝酸氧锆或硝酸锆。
根据本发明,优选地,所述载体改性剂前驱体选自la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属的水溶性盐。
根据本发明,优选地,所述活性助剂为选自re、co和ni中的至少一种金属的水溶性盐。
本发明中,上述氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体和活性助剂前驱体均可以在步骤(4)中的所述燃烧反应的条件下形成相应的金属氧化物。
本发明中,优选地,所述燃烧剂与所述混合溶液中金属总量的摩尔比为(0.7~3):1,优选为(1~2):1。所述混合溶液中金属可以为所述氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体和活性助剂前驱体中提供的金属。例如,所述氧化锆前驱体可以提供zr,所述载体改性剂前驱体可以提供la、ce、y、mn、ba、ca、mg、si和ti中的至少一种金属,所述氧化钼前驱体可以提供mo,所述活性助剂前驱体可以提供re、co和ni中的至少一种金属。
本发明中,所述燃烧剂可以为在步骤(3)的所述燃烧反应中自发燃烧而“烧掉”的化学物质。优选可以是燃点低于350~650℃的物质。优选地,所述燃烧剂为尿素、甘氨酸、乙二醇、氨基乙酸、甘油和甘露醇中的至少一种。
本发明中,优选地,所述稳定剂与所述混合溶液中金属总量的摩尔比为(0.01~0.2):1。
本发明中,优选地,所述稳定剂为柠檬酸、聚乙烯醇、二乙醇胺和乙酰丙酮中的至少一种。
本发明中,所述燃烧剂和稳定剂在经过步骤(3)的所述燃烧反应后均不残留于得到的耐硫甲烷化催化剂中。
根据本发明,得到所述混合溶液可以通过多种方法,只要能是氧化锆前驱体、载体改性剂前驱体、氧化钼前驱体、活性助剂前驱体、燃烧剂与稳定剂的完全溶解的溶液即可。优选地,步骤(1)中,得到所述混合溶液的过程包括:
(a)将氧化锆前驱体和载体改性剂前驱体在60~80℃恒温下溶解于去离子水中得到溶液a;
(b)将氧化钼前驱体和活性助剂前驱体在60~80℃下溶解于去离子水中得到溶液b;
(c)将溶液a和溶液b在60~80℃恒温下混合充分搅拌至澄清,再置于室温下得到溶液c;
(d)将所述燃烧剂和稳定剂加入溶液c,充分搅拌得到所述混合溶液。
本发明的步骤(2)中,得到所述透明胶状物的过程包括:将步骤(1)所得混合溶液在60℃水浴中进行浓缩2~4h,得到粘度为1000~3000mpa·s的所述透明胶状物。本发明中,该粘度为60℃下测得的粘度,可以采用brookfield公司的brookfielddv-ii+pro型旋转粘度计进行测量。
本发明第三方面,提供一种甲烷化方法,该方法包括:
(a)将本发明的耐硫甲烷化催化剂在含硫还原气氛下,在温度350~450℃、表压压力0.1~0.2mpa下进行预硫化反应2~6h,相对于1g的所述耐硫甲烷化催化剂,所述含硫还原气氛的流量为3~6l/h,所述含硫还原气氛含有硫化氢与氢气,所述含硫还原气氛中硫化氢含量为2~5体积%;
(b)在经步骤(a)得到的预硫化催化剂存在下,将含有氢气、一氧化碳、二氧化碳和硫化氢的混合气进行甲烷化反应,甲烷化反应温度为300~650℃,优选为400~600℃;甲烷化反应压力为0.5~6mpa;所述混合气中,氢气:二氧化碳:一氧化碳的体积比为(0.7~4):(0.5~2):1,硫化氢含量为0.4~0.8体积%,所述混合气的进气体积空速为5000~20000h-1。
以下将通过实施例对本发明进行详细描述。
以下实施例和对比例中,透明胶状物的粘度通过brookfield公司的brookfielddv-ii+pro型旋转粘度计测定。
对比例1
按照cn103433026a中实施例1的方法,
将2.278g的la(no3)3·6h2o置于10g去离子水中,经充分搅拌成浸渍液a;
称取10.0g预先经过干燥的单斜相商业zro2载体(alpha,比表面积91m2/g),将之放入浸渍液a中,剧烈搅拌2h,形成均匀的悬浮液后,用旋转蒸发仪蒸干其水分,再放入110℃干燥箱中烘干12h,再在600℃马弗炉中焙烧4h,得到表面负载la2o3的单斜相zro2;
称取3.680g的七钼酸铵((nh4)6mo7o24·4h2o)和0.474g的铼酸铵(nh4reo4)置于15g去离子水中,经搅拌配置成浸渍液b;将表面负载la2o3的单斜相zro2加入浸渍液b中,剧烈搅拌2h,形成均匀的悬浮液后,用旋转蒸发仪蒸干其水分,再放入110℃干燥箱中烘干12h,再在600℃马弗炉中焙烧4h,得到催化剂d1。
对比例2
称取22.775g的zro(no3)2·2h2o和5.089g的y(no3)3·6h2o置于70g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.680g的七钼酸铵((nh4)6mo7o24·4h2o)溶于5.5g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入10.373g的尿素和1.1g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃的水浴中进行浓缩至粘稠状,得到粘度约为2500mpa·s的透明胶状物;
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度450℃的马弗炉中,进行燃烧反应1h,冷却后经研磨过筛,得到催化剂d2。
对比例3
按照cn105879854a中实施例1的方法制备催化剂,
称取19.521g的zro(no3)2·2h2o和2.050g的la(no3)3·6h2o置于100g去离子水中,配置成溶液a;
称取3.312g的七钼酸铵((nh4)6mo7o24·4h2o)、17.657g的尿素和0.427g的铼酸铵(nh4reo4)溶于20g去离子水中得到溶液b;
然后将溶液a和b混合并充分搅拌后转入水热釜中,密闭水热釜后于160℃水热处理10h。
将水热后浆液抽滤并充分洗涤后,于120℃干燥脱水,600℃焙烧,得到催化剂d3。
实施例1
称取22.775g的zro(no3)2·2h2o和2.392g的la(no3)3·6h2o置于63g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.864g的七钼酸铵((nh4)6mo7o24·4h2o)和0.498g的铼酸铵(nh4reo4)溶于6.5g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入10.303g的尿素和1.1g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃的水浴中进行浓缩至粘稠状,得到粘度约为2010mpa·s的透明胶状物;
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度450℃的马弗炉中,进行燃烧反应1h,冷却后经研磨过筛,得到催化剂c1。
实施例2
称取20.346g的zro(no3)2·2h2o和2.233g的la(no3)3·6h2o置于56g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取4.121g的七钼酸铵((nh4)6mo7o24·4h2o)和0.465g的铼酸铵(nh4reo4)溶于7g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入7.658g的尿素和1.022g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃水浴中进行浓缩至粘稠状,得到粘度约为1820mpa·s的透明胶状物;
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度为500℃的马弗炉中,进行燃烧反应3h,冷却后经研磨过筛,得到催化剂c2。
实施例3
称取24.987g的zro(no3)2·2h2o和2.126g的la(no3)3·6h2o置于68g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.925g的七钼酸铵((nh4)6mo7o24·4h2o)和0.532g的铼酸铵(nh4reo4)溶于7g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入13.242g的尿素和和1.178g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃水浴中进行浓缩至粘稠状,得到粘度约为2190mpa·s的透明胶状物;
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度为400℃的马弗炉中,进行燃烧反应0.5h,冷却后经研磨过筛,得到催化剂c3。
实施例4
称取28.615g的zro(no3)2·2h2o和2.392g的la(no3)3·6h2o置于65g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.312g的七钼酸铵((nh4)6mo7o24·4h2o)和0.498g的铼酸铵(nh4reo4)溶于6g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入10.350g的尿素和1.105g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃水浴中进行浓缩至粘稠状,得到粘度约为2480mpa·s的透明胶状物。
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度为450℃的马弗炉中,进行燃烧反应1h,冷却后经研磨过筛,得到催化剂c4。
实施例5
称取23.10g的zro(no3)2·2h2o和2.392g的la(no3)3·6h2o置于63g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.864g的七钼酸铵((nh4)6mo7o24·4h2o)和1.088g的co(no3)2·6h2o溶于8g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将溶液b快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入13.240g的甘氨酸和1.10g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃水浴中进行浓缩至粘稠状,得到粘度约为2280mpa·s的透明胶状物。
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度为550℃的马弗炉中,进行燃烧反应1h,冷却后经研磨过筛,得到催化剂c5。
实施例6
按照实施例1的方法制备催化剂,不同的是,将“2.392g的la(no3)3·6h2o”替换为“2.270g的ce(no3)3·6h2o”,得到催化剂c6。
实施例7
称取23.751g的zro(no3)2·2h2o和1.595g的la(no3)3·6h2o置于63g去离子水中,在80℃水浴中经充分搅拌配置成溶液a;
称取3.864g的七钼酸铵((nh4)6mo7o24·4h2o)和0.332g的铼酸铵(nh4reo4)溶于6g去离子水中得到溶液b;
保持溶液a在80℃恒温水浴中,将b溶液快速加入到溶液a中,剧烈搅拌至溶液澄清,得到溶液c;
向溶液c中加入10.410g的尿素和和1.111g的柠檬酸,充分搅拌至完全溶解,得到混合溶液;
将混合溶液保持在60℃水浴中浓缩至粘稠状,得到粘度约为1860mpa·s的透明胶状物。
将透明胶状物转移至陶瓷坩埚中,置于已达到设定温度为450℃的马弗炉中,进行燃烧反应1h,冷却后经研磨过筛,得到催化剂c7。
实施例8
按照实施例1的方法制备催化剂,不同的是,将马弗炉的设定温度“450℃”替换为“350℃”,得到催化剂c8。
实施例9
按照实施例1的方法制备催化剂,不同的是,将“450℃的马弗炉中,进行燃烧反应1h”替换为“650℃的马弗炉中,进行燃烧反应3h”,得到催化剂c9。
实施例10
按照实施例1的方法制备催化剂,不同的是,将“10.303g的尿素”替换为“5.495g的尿素”,得到催化剂c10。
测试例1
利用rigaku公司d/max-2600/pc型x-射线衍射仪测定反应催化剂的晶相结构。
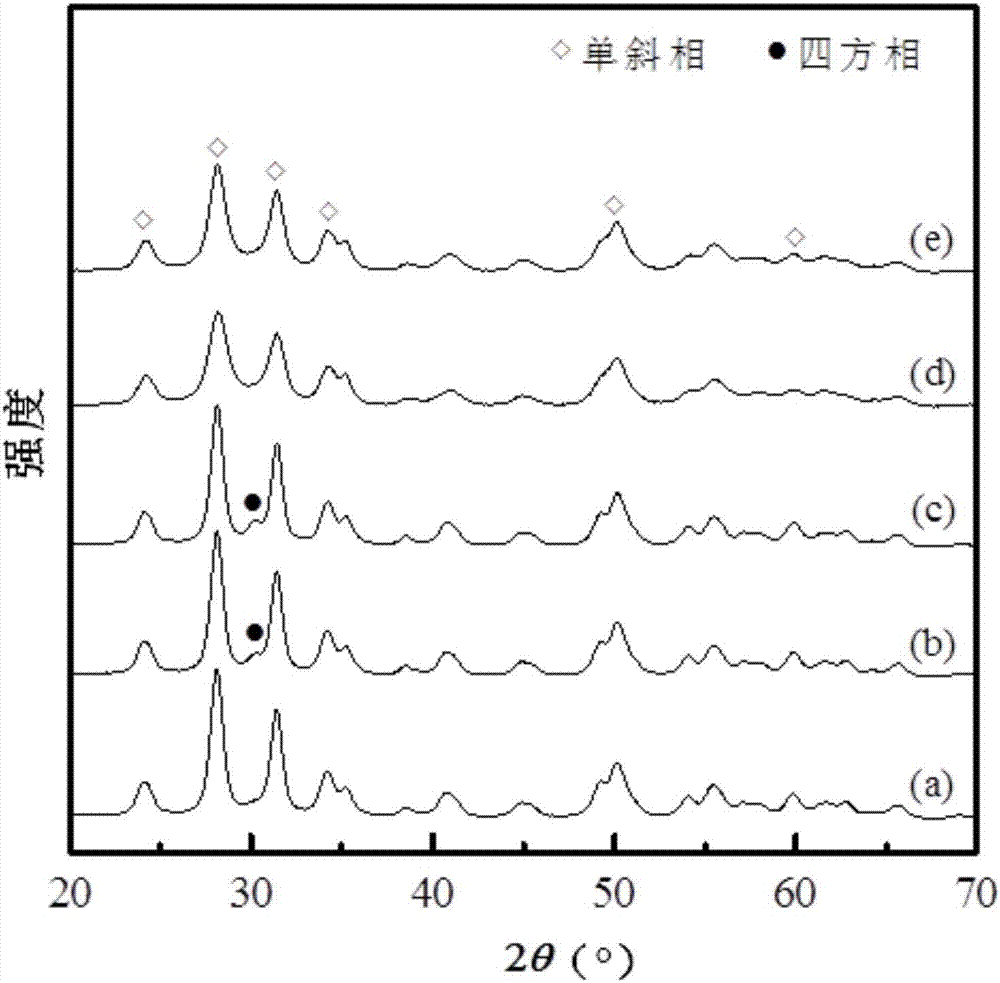
对比例1-3和实施例1-2所制备的催化剂的xrd谱图如图1所示(其中,a:d1,b:d2,c:d3,d:c1;e:c2)。可以看到,实施例1和实施例2的催化剂载体以单斜相zro2为主。而对比例中,对比例2和对比例3的催化剂载体中观察到有四方相的zro2晶型。此外,从图1中还可以看出本发明中催化剂的xrd谱图中没有mo物种的衍射峰出现,这表明mo物种高度分散在载体表面。实施例3-10的催化剂的xrd谱图与实施例1和实施例2的催化剂的xrd谱图类似。
测试例2
利用novananosem450扫描电子显微镜分析催化剂的表面形貌。图2、图3和图4分别是实施例1、实施例2和实施例9所制备催化剂的sem表征图。由图2和图3可以看出,催化剂表面由小粒子紧密堆积而成,粒子尺寸均一,表面较为粗糙,孔道丰富,这有利于活性组分的分散,增加催化剂表面活性位。图4是经650℃燃烧所制备催化剂的电镜图片,过高温度下,该催化剂表面致密平整,比表面积较低,不利于活性组分在表面的分散。
测试例3
利用jem-arm200f透射电子显微镜分析催化剂预硫化后活性组分的分散状态。
mo基耐硫甲烷化催化剂的主要活性相是mos2,催化剂在进行反应前需先预硫化,硫化后活性相mos2的形貌和分散状态对其活性具有至关重要的影响。图5和图6是实施例1和实施例2所制备催化剂经预硫化后的tem表征图。从图片中可以明显观察到典型的层状晶体结构,其晶格条纹的层间距为0.61nm左右,与mos2的(002)晶面间距一致。从图5和图6中均可以观察到mos2晶型的层状结构,且分散较为均匀,这有助于提供更多的催化活性位点。
测试例4
测试实施例c1-c10、对比例d1-d3所制备催化剂的甲烷化性能。
催化剂在反应前需先进行硫化,具体硫化条件为:将1g催化剂装填入固定床反应器中,通入4l/(g催化剂·h)流量的还原气(3体积%h2s/h2),以5℃/min的速率升温至400℃,在常压下硫化4h,结束后在还原气氛中调温至反应温度。
催化剂的反应条件为:原料气组成h2/co/co2=35/35/30(体积比),气体中h2s体积分数为0.6%,反应混合气的流速为6l/(g催化剂·h),设定反应温度为450℃,反应压力为3mpa。产物经脱硫和冷凝除水后进入安捷伦7890a型气相色谱仪进行在线检测,采用常规方法测定或计算甲烷化反应的co转化率。
实施例c1-c10和对比例d1-d3所制备催化剂的反应结果见表1。
表1
由表1的结果可以看出,本发明的催化剂比表面积均在150m2/g以上,其中催化剂c1-c6的比表面积均高于165m2/g。
与现有技术相比(d1-d3),本发明方法制备的耐硫甲烷化催化剂(特别是c1-c6)表现出优异的反应活性和稳定性,经100h反应后仍能保持较高的甲烷化转化率。对比例1、3中分别使用了现有技术的制备催化剂的方法,在与实施例1的配方相同的情况下,得到的d1比表面积差,得到的d3中zro2还含有单斜相之外的四方相,即催化剂载体晶型不纯,从而在表1得到的反应数据中,co转化率差。
比较实施例1(c1)与实施例7(c7)可以看出,控制催化活性组分钼、活性助剂、载体与载体改性剂的摩尔比在优选范围内能够获得性能更佳的催化剂。
比较实施例1(c1)与实施例8(c8)和实施例9(c9)可以看出,燃烧温度对催化剂的比表面积以及反应性能影响较大。燃烧温度过低,虽然产物可以获得较高的比表面积,但由于温度低会造成原料分解缓慢或分解不完全,影响最终催化剂的晶相结构,造成催化剂活性与稳定性下降;燃烧温度过高,燃烧时间长,会导致所制备催化剂比表面积降低,不利于活性组分的分散,从而导致较低的催化活性,采用优选的燃烧温度和燃烧时间能够获得性能更佳的催化剂。
比较实施例1(c1)与实施例10(c10)可以看出,燃烧剂用量不足时,产物的比表面积和反应活性相对较低,这可能与贫燃状况下燃烧反应不够充分有关。
测试例5
在不同co2含量的反应气氛中,比较实施例c1与对比例d1所制备催化剂的催化性能。
催化剂在反应前需先进行硫化,具体硫化条件如测试例4所述。
催化剂的反应条件:保持原料气中h2/co体积比为1,改变原料气中co2体积分数分别为30%,40%,50%,其余条件如测试例4的反应条件所述。
实施例c1-c3和对比例d1-d3所制备催化剂在不同co2浓度下进行耐硫甲烷化反应,反应2h后的co转化率见表2。
表2
由表2的结果可以看出,随着反应原料气中co2浓度提高,受热力学平衡限制,co转化率逐渐降低。但是,与现有技术制备的催化剂相比,本发明方法制备的耐硫甲烷化催化剂c1-c3在不同co2浓度下且高co2浓度下的耐硫甲烷化反应中均表现出更加优异的反应活性。
本发明所提供的耐硫甲烷化催化剂组成中含有活性助剂,并结合燃烧法一步合成,催化剂各组分前驱体在进行燃烧前可以以分子或原子水平均匀混合,有利于活性组分的分散;燃烧过程中高温时间较短,对催化剂孔结构破坏较小,使得最终产品可以获得较高的比表面积,同时可以抑制高温烧结所导致的活性中心数减少;活性助剂的添加可改善钼的分散性能和还原性能,有利于催化剂活性的提高;载体改性剂的添加一方面可以提高载体稳定性,抑制载体的相变与烧结,另一方面可以促进活性组分在载体表面的分散。
本发明所制备的催化剂具有较高的比表面积,活性组分分散较佳,在低温、高co2条件下表现出较优的甲烷化活性和稳定性。
以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!