一种兽药中喹诺酮类药物残留的检测方法与流程
本发明涉及分析化学技术领域,尤其是一种兽药中喹诺酮类药物残留的检测方法。
背景技术:
喹诺酮(4-quinolones),又称喹诺酮类、吡酮酸类或吡啶酮酸类,是一类人工合成的含4-喹诺酮基本结构,对细菌DNA螺旋酶具有选择性抑制的抗菌剂。由于喹诺酮既不是微生物分泌物又不是微生物分泌物的类似物,所以严格来说其只是人工合成的抗菌剂(Antibacterial)。
喹诺酮类抗生素是一类人畜通用的药物。因其具有抗菌谱广、抗菌活性强、与其他抗菌药物无交叉耐药性和毒副作用小等特点,被广泛应用于畜牧、水产等养殖业中,包括在鸡、鸭、鹅、猪、牛、羊、鱼、虾、蟹等的养殖中用于疾病防治。由于喹诺酮类药物在动物机体组织中的残留,人食用动物组织后喹诺酮类抗生素就在人体内残留蓄积,造成人体疾病对该药物的严重耐药性,影响人体疾病的治疗。人类长期食用含较低浓度喹诺酮类药物的动物性食品容易诱导耐药性的传递,从而影响该类药物的临床疗效。因此,喹诺酮类药物残留问题越来越引起人们的关注。联合国粮农组织/世界卫生组织食品添加剂专家联席委员会、欧盟都已制定了多种喹诺酮类药物在动物组织中的最高残留限量。美国FDA于2005年宣布禁止用于治疗家禽细菌感染的抗菌药物恩诺沙星的销售和使用。我国也于2002年规定了环丙沙星、单诺沙星、恩诺沙星、沙拉沙星、二氟沙星、恶喹酸和氟甲喹等7种喹诺酮类药物在动物肌肉组织中的最高残留限量为10~500μg/kg。
技术实现要素:
本发明所要解决的技术问题在于提供一种兽药中喹诺酮类药物残留的检测方法。
为解决上述技术问题,本发明的技术方案是:
一种兽药中喹诺酮类药物残留的检测方法,用液相色谱-串联质谱法测定兽药中的喹诺酮类药物残留,具体步骤如下:
(1)标准工作液和样品溶液在设定的液相色谱-串联质谱条件下进样,以质量浓度X为横坐标,峰面积的比值Y为纵坐标,绘制5点标准工作曲线,所述标准工作液为氧氟沙星、诺氟沙星、环丙沙星和恩诺沙星4种喹诺酮类药物混合工作液;
用标准工作曲线对样品进行定量,样品溶液中药物的响应值应在仪器检测的线性范围内,其中,所设定的液相色谱-串联质谱条件如下:
①液相色谱条件
色谱柱:Hypersil Gold C18,100mm×2.1mm,1.9μm
流动相:0.1%甲酸/甲醇;流速:0.35mL/min 柱温:35℃进样量10μL
液相梯度条件
②质谱条件
离子源:H-ESI源 扫描方式:负离子扫描;
检测方式:多反应检测 喷雾电压:3500V
雾化温度:400℃ 鞘气压力:35arb
辅气压力:10arb 毛细管温度:350℃
四种喹诺酮类药物的色谱保留时间和定性定量离子参数
(2)在上述色谱条件下,判断样品中是否存在相应的被测物,需要满足如下条件:样品溶液中出现的质量色谱峰保留时间与混合基质标准工作液一致,允许偏差小于±2.5%,该色谱峰所对应的药物在设定质谱定性离子的相对丰度与浓度相当的基质标准工作液的相对离子丰度一致,相对丰度偏差不超过设定 的规定,则可确定含有该药物。
所述5点标准工作曲线的绘制方法如下:从冰箱中取出100ng/mL的喹诺酮类药物混合标准工作液,恢复至室温后用0.1%甲酸稀释成浓度系列为0.5ng/ml、1ng/ml、2ng/ml、4ng/ml、8ng/ml的标准工作液,每一浓度进样3针,按其所得峰面积比值平均数Y为纵坐标,相应的标准品浓度X为横坐标绘制标准曲线。
上述兽药中喹诺酮类药物残留的检测方法,所述样品溶液的制备方法为试样经2%甲酸乙腈提取,氮吹后用0.1%甲酸复溶,过0.22μm微孔滤膜,试剂为色谱纯试剂。
上述兽药中喹诺酮药物残留的检测方法,所述样品溶液的制备方法,具体步骤如下:
(1)准确称取1g试样,精确到0.01g,置于50mL聚苯乙烯离心管中,加5mL 2%甲酸乙腈,涡动1-3min,离心力4000g-6000g,室温条件下离心10min,提取上清液1ml,55-60℃氮吹至干,以1mL0.1%甲酸复溶,复溶后再次用0.1%甲酸稀释10倍,最终溶液过0.22μm微孔滤膜,即可供液相色谱-串联质谱仪测定。
本发明的有益效果是:
上述兽药中喹诺酮药物残留的检测方法,用液相色谱-串联质谱法测定兽药中的喹诺酮类药物残留,该方法样品前处理时省去了净化过程,具有处理程序简单,快速,测定准确率高等特点;从兽药源头进行分析,有效保障养殖业用药安全,提高动物食品安全。
附图说明
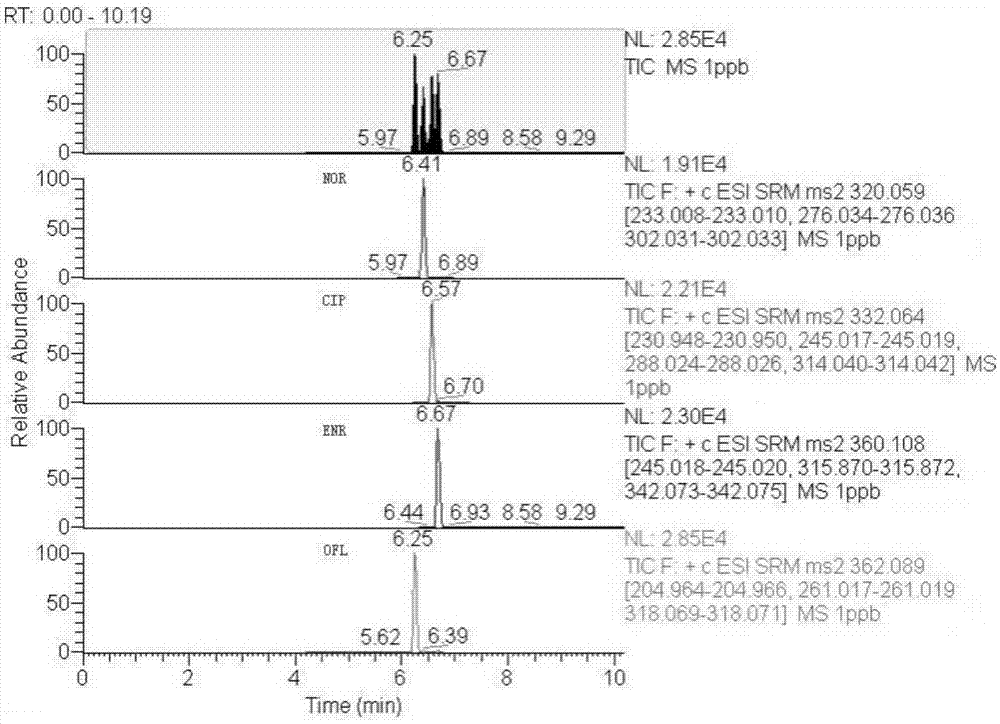
图1为兽药阴性样品检测喹诺酮类药物(四种沙星)总离子流图;
图2为兽药样品添加喹诺酮类药物(四种沙星)后总离子流图(药物浓度为1μg/kg)。
具体实施方式
为了使本领域的技术人员更好的理解本发明的技术方案,下面结合具体实施方式对本发明所述技术方案作进一步的详细说明。
实施例1
一、提取
选取4种不同的兽药样品做3种不同浓度的添加回收实验进行方法验证,兽药中待测药物的提取溶剂,分别采用乙腈、2%甲酸乙腈、甲醇和乙酸乙酯,4种试剂进行提取条件的选择。实验结果表明,对于兽药中的喹诺酮类兽药残留,2%甲酸乙腈的提取效率好于另外两种试剂。
具体操作:准确称取1g试样,精确到0.01g,置于50mL聚苯乙烯离心管中,加5mL 2%甲酸乙腈,涡动1-3min,离心力4000g-6000g,室温条件下离心10min,提取上清液1ml,55-60℃氮吹至干,以1mL0.1%甲酸复溶,复溶后再次用0.1%甲酸稀释10倍,最终溶液过0.22μm微孔滤膜,即可供液相色谱-串联质谱仪测定。
二、测定
本实验定量方法为外标法定量。从冰箱中分别取出100ng/mL的四种沙星药物混合标准工作液,用恢复至室温后用0.1%甲酸稀释成浓度系列为0.5ng/ml、1ng/ml、2ng/ml、4ng/ml、8ng/ml的标准工作液,每一浓度进样3针,按其所得峰面积比值平均数Y为纵坐标,相应的标准品浓度X为横坐标绘制标准曲线。根据试样中被测物的药物含量,选取响应值相近的药物标准工作液同时进行分析。标准工作液和待测液中药物的响应值均应在仪器线性响应范围内。如果含量超 过标准曲线范围,应用基质空白溶液(0.1%甲酸)稀释到合适浓度后分析。
①液相色谱条件
色谱柱:Hypersil Gold C18,100mm×2.1mm,1.9μm
流动相:0.1%甲酸/甲醇;流速:0.35mL/min 柱温:35℃进样量10μL
液相梯度条件
②质谱条件
离子源:H-ESI源 扫描方式:负离子扫描;
检测方式:多反应检测 喷雾电压:3500V
雾化温度:400℃ 鞘气压力:35arb
辅气压力:10arb 毛细管温度:350℃
四种喹诺酮类药物的色谱保留时间和定性定量离子参数
在上述色谱条件下,判断样品中是否存在相应的被测物,需要满足如下条件:样品溶液中出现的质量色谱峰保留时间与混合基质标准工作液一致,允许偏差小于±2.5%,该色谱峰所对应的药物在设定质谱定性离子的相对丰度与浓度相当的基质标准工作液的相对离子丰度一致,相对丰度偏差不超过设定的规定,则可确定含有该药物(如图1、图2所示)。
三、具体实施案例结果
本实验共选取4种兽药进行喹诺酮类药物做添加回收试验,其标准的曲线方程、回收率和精密度结果如下表:
表1四种喹诺酮类药物的标准曲线方程和相关系数
表2阿莫西林可溶性粉回收率测定结果
表3硫氰酸红霉素可溶性粉回收率测定结果
表4替米考星溶液回收率测定结果
表5地克珠利溶液回收率测定结果
表6阿莫西林可溶性粉精密度测定结果
表7硫氰酸红霉素可溶性粉精密度测定结果
表8替米考星溶液精密度测定结果
表9地克珠利溶液精密度测定结果
由以上案例可见,本发明采用液相色谱-串联质谱法测定兽药中的喹诺酮类药物残留,所述样品溶液的制备方法为试样首先通过水溶过滤,2%甲酸乙腈提取,氮吹后用0.1%甲酸复溶,过0.22μm微孔滤膜,然后用液相色谱-串联质谱法进行测定,外表法定量,回收率为62.3%~95.3%;样品批间和批内相对标准偏差均在10%左右。在样品提取和LC-MS/MS检测时,对样品提取液和检测条件都进行了优化处理。本发明的检测方法样品处理程序简单、快速,省去了样品净化过程,降低了检测成本,并且可快速准确的测定出兽药中残留的喹诺酮类药物。方法测定底限为50μg/kg,完全满足药物残留检测的要求。
上述参照具体实施方式对该一种兽药中喹诺酮类药物残留的检测方法进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!