一种乙烯基三氮唑产物的快速分离检测方法与流程
本发明涉及一种化合物分离检测方法,具体涉及一种乙烯基三氮唑产物的快速分离检测方法,确切的说是将利用多步溶胀法制备的多孔聚甲基丙烯酸缩水甘油酯微球与重氮树脂进行光交联,制备出重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球,以此为色谱柱填料,采用高效液相色谱(HPLC)使得乙烯基三氮唑与副产物在较短的时间内分离开。
背景技术:
乙烯基三氮唑作为一种带有碳碳双键水溶性的化合物,能够通过聚合反应得到带有富氮杂环的聚合物。该聚合物的物理化学性能,引起了热门的广泛关注,有很大的潜力被应用到在离子液体,燃料电池,有机合成,药物,医学等领域。因此,这种单体的发展会带动很多领域的进步。
乙烯基三氮唑的制备多是以乙酸乙烯酯与三氮唑的消去反应所制备而得。如Hideharu Mori等人在《Macromolecular Chemistry&Physics》杂志,2012,213,1803–1814报道了乙酸乙烯酯与三氮唑在氮气环境下通过消去反应得到了乙烯基三氮唑的单体。在中国专利CN103333127A中,在乙酸乙烯酯与三氮唑基础上,以醋酸汞为催化剂,醋酸汞的引入使得乙烯基三氮唑的产率增加到74%。在乙烯基三氮唑的合成过程会有醋酸的副产物的产生,其产物的纯度严重影响其应用,并且对乙烯基三氮唑产物进行有效分离检测的手段比较少。
气相色谱是对乙烯基三氮唑产物进行分离研究的常用手段,但其精度较低。另外,气相色谱在工作过程中温度都较高,特别是在使用火焰离子化检测仪,需要氢气来维持温度,使得整个检测过程有一定的危险性。且检测完成后样品得以破坏,很难被回收。因此,研究一种准确、快速、低耗的乙烯基三氮唑产物分离检测方法具有重要意义。
液相色谱分离技术是目前化合物产物分离检测中应用最为广泛的方法,具有快速、灵敏、高效的特点。且检测条件多在室温下,样品分离后均可回收,是一种很好的检测工具。但采用液相色谱分离时,需要一种特定的色谱柱填料,对乙烯基三氮唑产物进行有效分离的色谱柱至今未见报道。因此,这也提出了对一种能够用于乙烯基三氮唑产物进行高效液相色谱分离检测色谱柱填料的需求。
技术实现要素:
为此,本发明所要解决的技术问题在于在现有气相色谱精度低、安全性差,样品不易于回收,且在现有报道中并没有一种用高效液相色谱分离乙烯基三氮唑产物的方法,进而提出了一种乙烯基三氮唑产物的快速分离检测方法,以重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球为色谱柱填料制备色谱柱,采用HPLC实现对乙烯基三氮唑产物的快速分离。
为解决上述技术问题,本发明采用如下技术方案:
一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、由三氮唑和醋酸乙烯酯经消去反应所得到的产物经减压蒸馏、萃取、蒸馏浓缩后得到乙烯基三氮唑产物,取30-50微升产物加入到3-5ml体积比为(65-75):(25-35)的乙腈和水的混合液中,得到样品待测液;
S2、HPLC分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球分散于异丙醇中,以甲醇和异丙醇的混合液为加压液,通过装柱机装填到色谱柱中,以乙腈和水的混合液作为流动相,在温度为20~30℃、流速为0.2~1ml/min条件下,对乙烯基三氮唑产物样品待测液进行色谱分离,并采用高效液相色谱为检测工具对乙烯基三氮唑产物进行分离检测。
所述多孔聚甲基丙烯酸缩水甘油酯微球由下述方法制备:
S3-1、将单分散聚苯乙烯种子微球分散于水中形成种子微球分散液,在温度为25~35℃条件下向所述种子微球分散液中加入致孔剂乳液,在转速为290~310转/min的条件下机械搅拌20~24h进行溶胀,得到第一溶胀物;所述致孔剂乳液包括水、乳化剂和致孔剂;
S3-2、向所述第一溶胀物中加入聚合前驱体乳液,在转速为290~310转/min的条件下机械搅拌18~24h,使所述聚合前驱体乳液将所述聚苯乙烯种子微球再次溶胀,得到第二溶胀物;所述聚合前驱体乳液包括水、乳化剂、聚合单体、交联剂和引发剂;
S3-3、向所述第二溶胀物中加入聚乙烯醇的水溶液,并升温至70~75℃条件下反应20~24h,溶胀物内的聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,去除液相后形成具有多孔的聚合物微球;所述液相包括致孔剂、未反应的单体和交联剂;
S3-4、将上述制备的多孔聚合物微球置于稀硫酸溶液中进行水解,将水解后聚合物微球置于重氮树脂溶液中,室温下避光搅拌10~12h进行光交联反应,离心分离后将产物水洗、干燥,置于紫外光下曝光处理30~40min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
所述步骤S3-1~S3-3中,乳化剂为十二烷基硫酸钠,致孔剂为体积比为(1.0~1.4):(1.0~1.4)的邻苯二甲酸二丁酯和甲苯混合物,聚合单体为甲基丙烯酸缩水甘油酯,交联剂为二乙烯基苯,引发剂为过氧化苯甲酰,水为去离子水。
所述步骤S3-1中每10ml种子微球分散液含有0.24~0.28g所述聚苯乙烯种子微球;每0.24~0.28g聚苯乙烯种子微球需用20~24ml含有2.0ml~2.8ml致孔剂的乳液,其中乳化剂含量为0.35~0.4wt%。
所述步骤S3-2中每0.24~0.28g的聚苯乙烯种子微球需要聚合物前驱体乳液30~35ml,其中聚合前驱体乳液中含有所述聚合单体为0.5~0.7m,引发剂为0.01~0.15g,交联剂为1.8~2.2ml,乳化剂为0.23~0.27wt%。
所述的步骤S3-3中,在转速为110~130转/min的机械搅拌下,再向所述第二溶胀物中加入平均聚合度为1750±50的聚乙烯醇水溶液;升温到70~75℃进行聚合反应20~24h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的所述多孔微球中的液相,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取去除。
所述每0.24~0.28g的聚苯乙烯种子微球所对应的聚乙烯醇水溶液加入量为2.7~3.3ml;所述聚乙烯醇水溶液质量分数为8~12wt%。
所述步骤S3-4中,应在转速为110~130转/min的机械搅拌下,将所制备的多孔聚合物微球加入到稀硫酸溶液中水解,水解温度为55~65℃,水解时间为12~15h;所述水解后的多孔聚合物微球在离心分离后加入到重氮树脂水溶液中。
所述步骤S3-4中使用的稀硫酸溶液为每30ml水中含有0.1~0.15mol硫酸,重氮树脂水溶液的浓度为10~20mg/mL。
所述步骤S2中,每0.24~0.28g的聚苯乙烯种子微球所制备的1.20~1.40g经重氮树脂修饰的多孔聚合物微球中加入的异丙醇的量为20~22ml;加压液中甲醇和异丙醇的体积比为(0.9-1.1):(0.9-1.1);流动相中乙腈和水的体积比为(65-75):(25-35)。
本发明的上述技术方案相比现有技术有以下优点:
本发明采用重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球作为色谱柱填料对乙烯基三氮唑产物进行分离,该填料以多步溶胀聚合所制备的较为均一多孔聚甲基丙烯酸缩水甘油酯微球为基础,再将聚合物微球水解出羟基后,与带有重氮基团的重氮树脂进行光交联反应制备得到。该色谱柱利用所制备色谱柱填料的疏水性减少对亲水乙烯基三氮唑产物的整体的保留时间,与此同时通过所修饰重氮树脂的上的仲氨基增强了对副产物乙酸等的保留能力,从而使得乙烯基三氮唑与副产物在较短的时间内分离开。实验结果表明,在流速为0.5ml/min,流动相为7:3体积比的乙腈和水混合液条件下进行HPLC分离,整个分离过程仅耗用不到5min,且其分离效果远好于C18的商品色谱柱,是一种非常有效的对乙烯基三氮唑产物进行分离的方法。
采用本方法采用高效液相色谱为分离工具,利用疏水性聚合物微球为色谱柱填料以减少具有亲水性乙烯基三氮唑产物的整体保留时间,从而达到对乙烯基三氮唑产物的快速分离检测,其分离检测快速、操作简单,且重现性好,有着较高的实用价值。
附图说明
图1为实施例1制备得到的多孔聚合物微球扫描电镜照片;
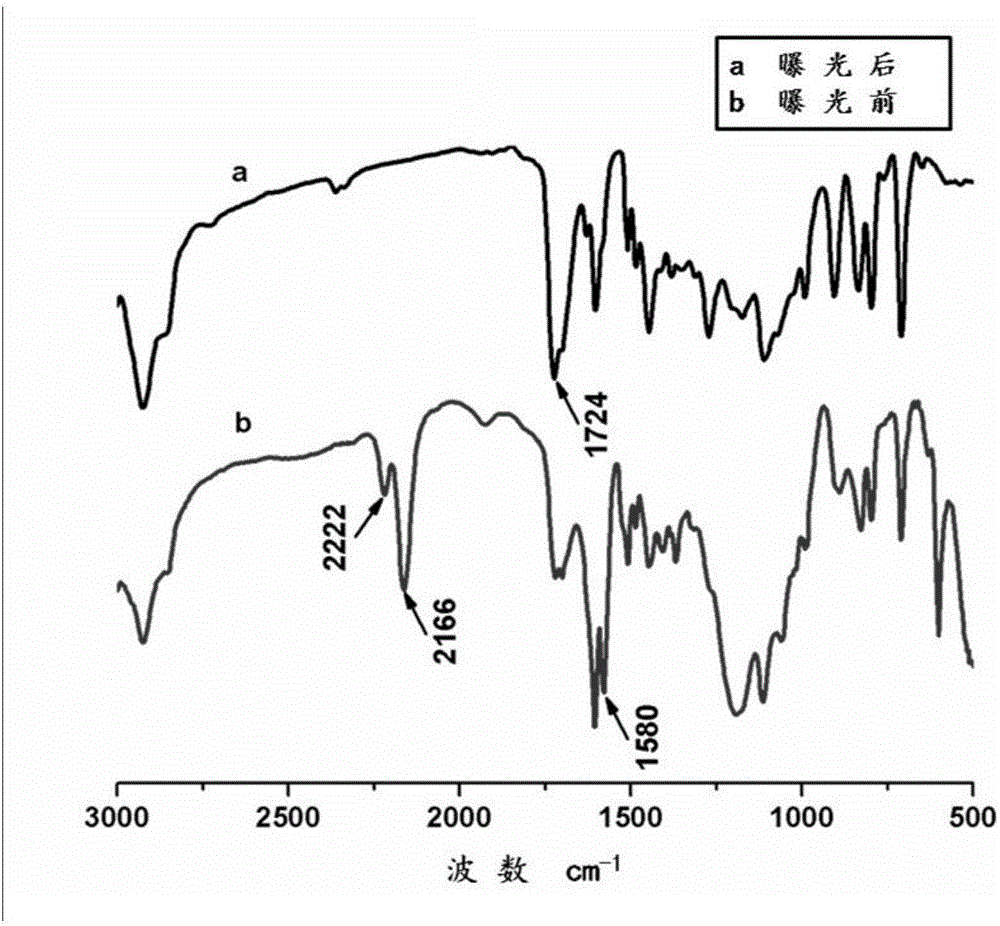
图2为实施例1制备的曝光前后多孔聚合物微球傅立叶变换红外光谱分析曲线;
图3为实施例1以重氮树脂修饰的多孔聚合物微球为色谱柱填料对疏水物质的分离效果;
图4为乙烯基三氮唑产物的分离效果图。
具体实施方式
为了使本发明的目的、技术方案和优点更加清楚,下面将对本发明的实施方式作进一步地详细描述。本发明可以以许多不同的形式实施,而不应该被理解为限于在此阐述的实施例。相反,提供这些实施例,使得本公开将是彻底和完整的,并且将把本发明的构思充分传达给本领域技术人员,本发明将仅由权利要求来限定。
实施例1-5中乙烯基三氮唑产物的制备方法为:由三氮唑和醋酸乙烯酯经消去反应所得到乙烯基三氮唑产物经减压蒸馏、萃取、蒸馏浓缩后而得,具体为:将150ml(1.62mol)的醋酸乙烯酯、0.2g的对苯二酚、9.5g的乙酸汞、9.6ml的三氟乙酸、37.0g(0.51mol)的1,2,4-三氮唑加入接有球形冷凝管的500ml的烧瓶中,并向其中加入磁子,在常温下强力电磁搅拌10-20min,油浴加热,控制反应温度为70℃。反应在氮气保护下进行。当反应3h之后,停止加热和搅拌,静置冷却,此时溶液呈深褐色,对烧瓶中的溶液进行减压蒸馏,将未反应的醋酸乙烯酯减压蒸出。用600ml的饱和碳酸氢钠溶液中和烧瓶中的剩余液体,直至pH值接近7。将中和后的液体加入分液漏斗中,并在其中加入一定量的乙醚,进行多次萃取,收集上层液体,进行旋转蒸发,将其中的溶剂乙醚蒸出。用无水硫酸钠将烧瓶中剩余的液体干燥,对干燥后的液体进行减压蒸馏,收集90℃/25mmHg,馏分,即为乙烯基三氮唑产物,溶液为无色透明的液体
作为另一种选择,所述的乙烯基三氮唑产物也可以是按照中国专利CN103333127A提供的方法制备出乙烯基三氮唑产物。
实施例1:
本实施例一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、取40微升产物混入到3ml体积比为70:30的乙腈和水的混合液中,得到样品待测液;
S2、HPL C分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球1.25g分散于20ml异丙醇中,通过以体积为1:1的甲醇和异丙醇的混合液为加压液,通过装柱机装填到高度为75mm,直径为4.6mm的不锈钢色谱柱中,并在流速为0.5ml/min对乙烯基三氮唑产物样品待测液进行了分离。如图4所示,曲线a为本实施例所制备的经重氮树脂修饰改性的多孔微球对乙烯基三氮唑产物的分离效果,结果表明:改性后的微球作为色谱柱填料具有良好的分离效果,其中波峰依次为乙烯基三氮唑、副产物醋酸;曲线b为多孔微球改性之前对乙烯基三氮唑产物的分离效果,结果表明:未改性的微球作为色谱柱填料并没有将乙烯基三氮唑产物分开;曲线c为商品化C18色谱柱对乙烯基三氮唑产物的分离效果,结果表明:商品化C18色谱柱也并没有将乙烯基三氮唑产物很好地分离。
其中重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的制备方法如下:
S3-1、在30℃下,将10ml含有0.26g聚苯乙烯种子微球的分散液加入到含有1.2ml邻苯二甲酸二丁酯和1.2ml甲苯组成的致孔剂的20ml致孔剂乳液中,其中乳化剂十二烷基硫酸钠含量为0.375wt%;在转速为300转/min的机械搅拌下溶胀24h,使得聚苯乙烯种子微球完全被邻苯二甲酸二丁酯和甲苯混合液溶胀,得到第一溶胀物。
S3-2、将30ml含有聚合单体甲基丙烯酸缩水甘油酯0.6ml、引发剂过氧化苯甲酰0.12g、交联剂二乙烯基苯2.0ml的聚合前驱体乳液加入到所述第一溶胀物中,聚合前驱体乳液中乳化剂十二烷基硫酸钠含量为0.25wt%。在转速为300转/min的机械搅拌下溶胀24h,所述第一溶胀物被充分溶胀,得到第二溶胀物。
S3-3、在转速为120转/min的机械搅拌下,将质量分数为10%聚乙烯醇(平均聚合度为1750±50)水溶液3.0ml加入到所述第二溶胀物中,升温到70℃进行聚合反应24h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取液相后即得到多孔聚合物微球。
S3-4、在转速为120转/min的机械搅拌下,所制备的多孔聚合物微球加入到30ml含有0.1mol硫酸水溶液中,在温度为60℃下水解12h,将水解后的多孔聚合物微球离心分离后,加入到浓度为15mg/mL重氮树脂水溶液中在室温下避光搅拌12h,利用带有重氮基团的重氮树脂与水解后聚合物表面微球的羟基进行光交联反应,然后离心分离后将产物水洗、干燥,置于紫外光下曝光处理30min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
本实施例制备的经重氮树脂修饰的多孔聚合物微球的扫描电镜照片如图1所示,从图中可以看出所得到的多孔微球具有较好的均一性。
图2为本实施例所得的曝光前和曝光后的多孔聚合物微球的傅立叶变换红外光谱分析曲线,与曝光前相比,曝光后重氮基团所对应的2222cm-1与2166cm-1及重氮树脂上的苯环和羟基相互作用所产生的1580cm-1的红外吸收峰的消失,表明曝光后重氮树脂发生光交联反应,且1724cm-1增强是由于重氮基与羟基光交联后产生的新键产生而引起,与Tingbing Cao等人在杂志Journal of Physical Chemistry B,2001,48,11941-11944中所报道的波峰变化基本一致,说明重氮树脂修饰到了微球表面。
此外,本发明的发明人还对制备得到的重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的疏水性进行了研究,具体是以体积比为7:3的乙腈和水的混合液作为流动相,在0.5ml/min的流速下,对五种苯类似物进行了高效液相色谱分离,如附图3所示,改性后的微球作为色谱柱填料对疏水物质的具有良好的分离效果,其中波峰依次尿嘧啶、苯、丙苯、萘、芴。该实验表明所制备的微球有很好的疏水分离能力,能够有效的减少亲水性产物的保留时间。
实施例2:
本实施例一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、取50微升产物混入到3.5ml体积比为65:35的乙腈和水的混合液中,得到样品待测液;
S2、HPLC分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球1.40g分散于20ml异丙醇中,通过以体积为0.9:1.1的甲醇和异丙醇的混合液为加压液,通过装柱机装填到高度为75mm,直径为4.6mm的不锈钢色谱柱中,以体积比为65:35的乙腈和水的混合液作为流动相,在温度为25℃下,流速为0.6ml/min,对乙烯基三氮唑产物样品待测液进行了高效液相色谱分离,表现出很好的分离效果。
其中重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的制备方法如下:
S3-1、在32℃下,将10ml含有0.28g聚苯乙烯种子微球的分散液加入到含有1.1ml邻苯二甲酸二丁酯和1.2ml甲苯组成的致孔剂的20ml致孔剂乳液中,其中乳化剂十二烷基硫酸钠含量为0.360wt%;在转速为305转的机械搅拌下溶胀20h,使得聚苯乙烯种子微球完全被邻苯二甲酸二丁酯和甲苯混合液溶胀,得到第一溶胀物。
S3-2、将30ml含有聚合单体甲基丙烯酸缩水甘油酯0.5ml、引发剂过氧化苯甲酰0.12g、交联剂二乙烯基苯2.1ml的聚合前驱体乳液加入到所述第一溶胀物中,聚合前驱体乳液中乳化剂十二烷基硫酸钠含量为0.25wt%。在转速为300转/min的机械搅拌下溶胀24h,所述第一溶胀物被充分溶胀,得到第二溶胀物。
S3-3、在转速为120转/min的机械搅拌下,将质量分数为12%聚乙烯醇(平均聚合度为1750±50)水溶液3.3ml加入到所述第二溶胀物中,升温到75℃进行聚合反应20h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取液相后即得到多孔聚合物微球。
S3-4、在转速为120转/min的机械搅拌下,所制备的多孔聚合物微球加入到30ml含有0.12mol硫酸水溶液中,在温度为65℃下水解12h,将水解后的多孔聚合物微球离心分离后,加入到浓度为18mg/mL重氮树脂水溶液中在室温下避光搅拌10h,利用带有重氮基团的重氮树脂与水解后聚合物表面微球的羟基进行光交联反应,然后离心分离后将产物水洗、干燥,置于紫外光下曝光处理35min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
实施例3:
本实施例一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、取45微升产物混入到4.5ml体积比为75:25的乙腈和水的混合液中,得到样品待测液;
S2、HPLC分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球1.30g分散于22ml异丙醇中,通过以体积为1.1:0.9的甲醇和异丙醇的混合液为加压液,通过装柱机装填到度为75mm,直径为4.6mm的不锈钢色谱柱中,以体积比为75:25的乙腈和水的混合液作为流动相,在温度为30℃下,流速为0.8ml/min,对乙烯基三氮唑产物样品待测液进行了高效液相色谱分离,表现出很好的分离效果。
其中重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的制备方法如下:
S3-1、在28℃下,将10ml含有0.27g聚苯乙烯种子微球的分散液加入到含有1.3ml邻苯二甲酸二丁酯和1.1ml甲苯组成的致孔剂的20ml乳液中,其中乳化剂十二烷基硫酸钠含量为0.40wt%;在转速为295转的机械搅拌下溶胀22h,使得聚苯乙烯种子微球完全被邻苯二甲酸二丁酯和甲苯混合液溶胀,得到第一溶胀物。
S3-2、将30ml含有聚合单体甲基丙烯酸缩水甘油酯0.7ml、引发剂过氧化苯甲酰0.15g、交联剂二乙烯基苯1.9ml的聚合前驱体乳液加入到所述第一溶胀物中,聚合前驱体乳液中乳化剂十二烷基硫酸钠含量为0.26wt%。在转速为305转的机械搅拌下溶胀20h,所述第一溶胀物被充分溶胀,得到第二溶胀物。
S3-3、在转速为115转的机械搅拌下,将质量分数为8%聚乙烯醇(平均聚合度为1750±50)水溶液2.7ml加入到所述第二溶胀物中,升温到72℃进行聚合反应22h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取液相后即得到多孔微球。
S3-4、在转速为125转的机械搅拌下,所制备的多孔聚合物微球加入到30ml含有0.15mol硫酸水溶液中,在温度为58℃下水解15h,将水解后的多孔聚合物微球离心分离后,加入到浓度为20mg/mL重氮树脂水溶液中在室温下避光搅拌12h,利用带有重氮基团的重氮树脂与水解后聚合物表面微球的羟基进行光交联反应,然后离心分离后将产物水洗、干燥,置于紫外光下曝光处理35min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
实施例4:
本实施例一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、取35微升产物混入到体积比为3ml体积比为70:30的乙腈和水的混合液中,得到样品待测液;
S2、HPLC分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球1.20g分散于23ml异丙醇中,通过以体积为1:1的甲醇和异丙醇的混合液为加压液,通过装柱机装填到度为75mm,直径为4.6mm的不锈钢色谱柱中,以体积比为7:3的乙腈和水的混合液作为流动相,在温度为30℃下,流速为0.7ml/min,对乙烯基三氮唑产物样品待测液进行了高效液相色谱分离,表现出很好的分离效果。
其中重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的制备方法如下:
S3-1、在32℃下,将10ml含有0.27g聚苯乙烯种子微球的分散液加入到含有1.3ml邻苯二甲酸二丁酯和1.2ml甲苯组成的致孔剂的20ml乳液中,其中乳化剂十二烷基硫酸钠含量为0.36wt%;在转速为305转的机械搅拌下溶胀22h,使得聚苯乙烯种子微球完全被邻苯二甲酸二丁酯和甲苯混合液溶胀,得到第一溶胀物。
S3-2、将30ml含有聚合单体甲基丙烯酸缩水甘油酯0.7ml、引发剂过氧化苯甲酰0.12g、交联剂二乙烯基苯2.0 0ml的聚合前驱体乳液加入到所述第一溶胀物中,聚合前驱体乳液中乳化剂十二烷基硫酸钠含量为0.26wt%。在转速为305转的机械搅拌下溶胀20h,所述第一溶胀物被充分溶胀,得到第二溶胀物。
S3-3、在转速为125转的机械搅拌下,将质量分数为10%聚乙烯醇(平均聚合度为1750±50)水溶液2.8ml加入到所述第二溶胀物中,升温到72℃进行聚合反应22h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取液相后即得到多孔微球。
S3-4、在转速为125转的机械搅拌下,所制备的多孔聚合物微球加入到30ml含有0.12mol硫酸水溶液中,在温度为60℃下水解15h,将水解后的多孔聚合物微球离心分离后,加入到浓度为18mg/mL重氮树脂水溶液中在室温下避光搅拌12h,利用带有重氮基团的重氮树脂与水解后聚合物表面微球的羟基进行光交联反应,然后离心分离后将产物水洗、干燥,置于紫外光下曝光处理35min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
实施例5:
本实施例一种乙烯基三氮唑产物的快速分离检测方法,包括以下步骤:
S1、取40微升产物混入到4ml体积比为70:30的乙腈和水的混合液中,得到样品待测液;
S2、HPLC分离检测:将重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球1.35g分散于21ml异丙醇中,通过以体积为1:1的甲醇和异丙醇的混合液为加压液,通过装柱机装填到度为75mm,直径为4.6mm的不锈钢色谱柱中,以体积比为7:3的乙腈和水的混合液作为流动相,在温度为30℃下,流速为0.3ml/min,对乙烯基三氮唑产物样品待测液进行了高效液相色谱分离,表现出很好的分离效果。
其中重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球的制备方法如下:
S3-1、在35℃下,将10ml含有0.24g聚苯乙烯种子微球的分散液加入到含有1.2ml邻苯二甲酸二丁酯和1.1ml甲苯组成的致孔剂的22ml致孔剂乳液中,其中乳化剂十二烷基硫酸钠含量为0.35wt%;在转速为310转/min的机械搅拌下溶胀23h,使得聚苯乙烯种子微球完全被邻苯二甲酸二丁酯和甲苯混合液溶胀,得到第一溶胀物。
S3-2、将32ml含有聚合单体甲基丙烯酸缩水甘油酯0.5ml、引发剂过氧化苯甲酰0.13g、交联剂二乙烯基苯2.1ml的聚合前驱体乳液加入到所述第一溶胀物中,聚合前驱体乳液中乳化剂十二烷基硫酸钠含量为0.25wt%。在转速为305转的机械搅拌下溶胀20h,所述第一溶胀物被充分溶胀,得到第二溶胀物。
S3-3、在转速为125转的机械搅拌下,将质量分数为12%聚乙烯醇(平均聚合度为1750±50)水溶液3.1ml加入到所述第二溶胀物中,升温到74℃进行聚合反应20h,所述聚合单体与交联剂在引发剂作用下发生聚合反应形成包裹有液相的多孔微球,离心分离,将致孔剂、未反应的单体和交联剂用四氢呋喃萃取液相后即得到多孔聚合物微球。
S3-4、在转速为110转/min的机械搅拌下,所制备的多孔聚合物微球加入到30ml含有0.14mol硫酸水溶液中,在温度为58℃下水解13h,将水解后的多孔聚合物微球离心分离后,加入到浓度为18mg/mL重氮树脂水溶液中在室温下避光搅拌11h,利用带有重氮基团的重氮树脂与水解后聚合物表面微球的羟基进行光交联反应,然后离心分离后将产物水洗、干燥,置于紫外光下曝光处理40min,即可得到重氮树脂修饰的多孔聚甲基丙烯酸缩水甘油酯微球。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!