用于分子的跨膜递送的化合物和方法与流程
本发明涉及用于分子跨过生物膜递送入细胞(任选存在随后的细胞内捕获)的递送系统和相关方法。
背景
在许多医学病症的病因学和发病机制中,蛋白质病理学是常见的共同特征,从突变蛋白质的功能失常到病态的功能获得,其中特定蛋白质获得使其具有毒性的新性质。理论上讲,通过基因疗法抑制这些蛋白质的合成可为患有这样的蛋白质异常的患者带来希望。
近些年的主要进展之一是通过使用干扰小RNA(siRNA)的RNA干扰来沉默特定基因的概念。RNA干扰是基于短(≈19-27碱基对)的双链RNA序列,其能够与细胞生物系统(如裂解双链RNA以产生siRNA的Dicer蛋白复合物,以及RNA诱导沉默复合物(RISC)等)相呼应地发挥作用,以抑制翻译并为降解特定mRNA序列标记,由此在翻译阶段抑制基因表达。还建议使用反义寡核苷酸(ASO)(未修饰或化学修饰的DNA分子的短序列(通常13-25个核苷酸),其与特定mRNA互补)来抑制各个特定靶蛋白的表达并阻断其产生。
然而,尽管这样的用于医疗护理的方法有巨大的潜在益处,但是由于siRNA相对较大和高度带电荷的结构(平均MW:13kD,约40个带负电荷的磷酸酯基团),这样的大分子的跨膜递送仍然是显著挑战。因此,siRNA的跨膜递送需要克服非常大的能量屏障。
膜偶极电位是存在于任何磷脂膜,在水/膜界面和膜中心(里面为正)之间的电位。它被认为是由于高度有序的磷脂甘油基酯键的羰基所产生的。其振幅为约220-280mV。由于其在高度疏水的环境(介电常数为2-4)中,其转化为108-109V/m的非常强的电场。可以想到,膜偶极电位和相关的膜内电场对于膜蛋白的功能是非常重要的。偶极电位可能在决定膜蛋白的构象和活性中具有重要的生理作用。然而迄今为止,尚未将偶极电位用于医疗用途。
已开发多种方法用于寡核苷酸跨生物膜的递送。这些方法包括病毒载体和非病毒递送系统(如阳离子脂质或脂质体)。然而迄今为止,这些方法的使用大部分限于体外施用,或者用于体内局部给药(例如,通过向眼直接注射或向肺直接给药)。例如,电穿孔是用于大分子的跨生物屏障递送的有效并广泛使用的方法。根据该方法,向细胞悬浮液施加外电场,这导致带电荷的靶分子与膜的碰撞,随后暂时的和局部的膜失稳,并且因此大分子通过进入细胞。然而迄今为止,电穿孔主要用于体外。体内电穿孔具有有限的成功,并仅对于特定器官(如肌肉、肺)尝试,其中可将外部电极插入靶器官中。
总之,大分子(如寡核苷酸和其它治疗剂)通过细胞膜和其它生物屏障(如血脑屏障)的递送仍是显著的未满足的需求,并且这样的大分子的全身递送(即通过静脉给药或口服的递送)仍是巨大的未解决的挑战。
概述
本发明的实施方案提供递送系统,其基于新的、合理设计的“分子马达”。根据本发明的实施方案的“分子马达”可用于药物(其可包括小分子药物或大分子,如肽、蛋白质或寡核苷酸(例如单链或双链RNA或DNA))的跨膜递送。在具体实施方案中,大分子可包括用于基因沉默的RNA链(即siRNA(干扰小RNA))或设计用作反义寡核苷酸(ASO)的DNA序列。
根据本发明的实施方案的药物(如小分子药物或大分子)与“分子马达”的缀合物可在医疗实践中使用,如用于其中异常蛋白质或蛋白质功能障碍发挥作用以及其中沉默编码这些蛋白质的基因的表达可以是有益的医学病症的治疗;例如在退行性病症、癌症、毒性或缺血性损伤、感染或免疫介导的病症的治疗中。
在一个实施方案中提供用于药物的跨膜递送的化合物,所述化合物具有式II
(A)a-B-Q-L
(式II)
其中a是1、2、3或4的整数,并且其中A选自如式III、IV和V所示的结构:
其中M选自-O-或-CH2-;并且g和h各自为选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16的整数;
并且其中
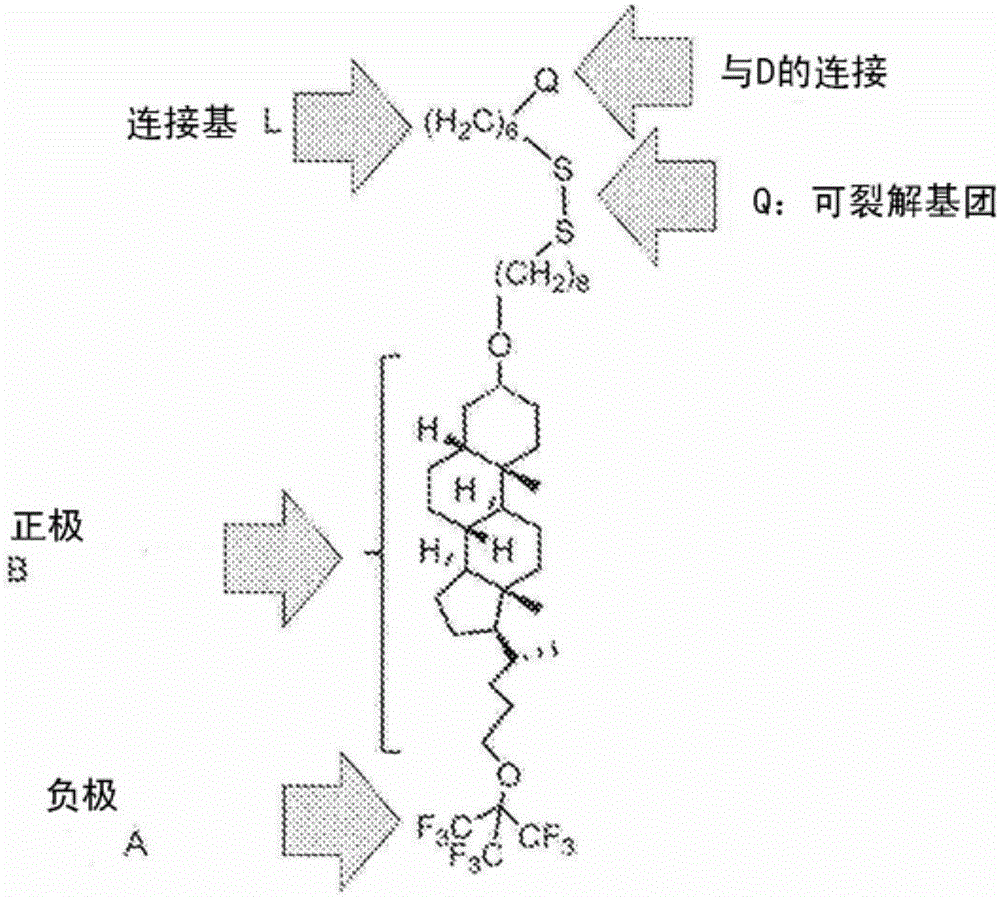
B是饱和或部分饱和的线性、支化或环状的C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、C19、C20、C21、C22、C23、C24、C25、C26、C27、C28、C29、C30、C31、C32、C33、C34、C35、C36、C37、C38、C39、C40、C41、C42烷基、亚烷基、亚杂烷基、芳基、杂芳基;甾体或其组合;
Q不存在或者选自酯、硫酯、酰胺、氨基甲酸酯、二硫键[-(S-S)-]、醚[-O-]、pH敏感基团和氧化还原敏感基团;
L不存在或者为任选被取代的线性、环状或支化的饱和、不饱和或部分饱和的C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、C19、C20、C21、C22、C23、C24、C25、C26、C27、C28、C29、C30、C31、C32、C33、C34、C35、C36、C37、C38、C39、C40、C41、C42烷基、亚烷基、亚杂烷基、芳基、杂芳基;甾体或-(O-CH2-CH2)u-,其中u为1、2、3、4、5、6、7、8、9、10的整数;或其组合;
所述化合物可连接至药物。
在一个实施方案中提供包含连接至药物的如上所述的化合物的缀合物。
本发明的一些实施方案涉及用于药物跨生物膜递送的方法,所述方法包括将细胞与如上所述的缀合物孵育。
另一实施方案涉及用于治疗医学病症的方法,所述方法包括向有需要的患者给药足够量的包含如上所述的缀合物的药物组合物。
附图简述
现会参照以下的说明性附图,以非限制性方式结合某些实施例和实施方案来描述本发明,从而可更全面地理解本发明。在附图中:
图1A是根据本发明的实施方案的化合物的活性基础的不对称极性的原理的图示呈现;
图1B图示描述如式IX的化合物所说明的本发明分子的基本结构;
图2图示说明根据本发明的实施方案的缀合物的可能的作用机制;
图3图示说明利用Dicer酶将siRNA捕获在细胞质中的机制;
图4表明包含本发明的“分子马达”,缀合至Cas9蛋白的缀合物的示例性结构;
图5A和5B显示根据本发明的实施方案的化合物的生物学性能的结果;图5A显示荧光显微术的结果,并且图5B显示孵育24小时后通过ELISA读取器的定量分析的结果。
发明详述
本发明的实施方案涉及新化合物,其可作为用于药物跨生物膜(例如磷脂细胞膜)进入细胞质的递送系统。本发明的实施方案的化合物包含新的、合理设计的“分子马达”,其被设计为在磷脂膜内,利用与膜偶极电位相关的内部膜电场从膜/水界面移动到膜核心。当连接至药物时,所述递送系统发挥将药物拉向膜核心,并便于跨膜移动的作用。例如,该递送系统被设计用于递送治疗性大分子:蛋白质或寡核苷酸(后者为单链或双链DNA或RNA)。例如,所述递送系统被设计用于递送反义寡核苷酸(ASO)、siRNA和治疗性蛋白质(如Cas9蛋白)。
根据本发明的实施方案的化合物的结构的原理之一是“不对称极性”的原理(其关于根据它们的logP为疏水、不带电荷的分子分配入生物膜(参见图1A))。所述分子是极性的,它们局部电荷不均匀地分布:局部负电荷高度局部化和集中,而局部正电荷在分子内沿烃链分散。另外,局部正电荷也通过与膜脂质环境内相邻的烃链的London型相互作用(London色散力)掩蔽。因此,如图1A中图示说明,根据本发明的实施方案的分子以带负电荷的分子的形式在膜环境内移动。由于膜内电场在膜/水界面处具有负极,在膜中心处具有正极,分子在相关电场中向膜中心移动,并且当其连接至负载物(cargo)(如siRNA或ASO、治疗性蛋白质或其它药物),它将所述负载物拉至膜中心。
另外,当在根据本发明的实施方案的分子中包含任选存在的可裂解基团(如二硫键基团(disulfide group)或寡核苷酸序列可通过Dicer酶裂解)时,所述可裂解基团可发挥在靶细胞的细胞质中捕获负载物(如siRNA或ASO或其它药物),并且还辅助维持缀合物跨细胞膜的浓度梯度的作用。因此在本发明的上下文中,术语“可裂解基团”涉及在某些生理条件(如pH变化或氧化还原状态变化)下,能够进行自发性或酶介导的裂解的化学基团。可裂解基团的实例为酯、硫酯、酰胺、氨基甲酸酯、二硫键[-(S-S)-]、醚(-O-)或硫醚(-S-)。理论上讲,可裂解基团可辅助药物在跨膜通过后在其靶细胞中药物的捕获,或辅助维持跨生物膜的浓度梯度。
例如,在根据本发明的实施方案的缀合物包含siRNA、ASO或治疗性蛋白质以及二硫键基团的情况中,一旦在细胞质内,普遍的周围还原性环境会发挥将所述二硫键还原为-SH基团,同时从递送基团释放负载物的作用。在无“分子马达”(递送基团)下,则负载物大分子会被捕获在细胞质中,其中例如,在siRNA的情况中,其会易于与RNA诱导沉默复合物(RISC)相互作用,以沉默特定基因的表达。根据本发明的实施方案,所述基因可编码参与特定疾病的病因学或发病机制的蛋白质。
根据本发明的一个实施方案,所述负载物是治疗性蛋白质,其被跨细胞膜递送以对细胞内蛋白质靶点发挥有益的治疗作用。
细胞内靶点的蛋白质类药物(PDIT)的领域是部分源自人基因组序列测定工程的完成的新领域,其使得能够鉴定大量用于潜在的医学干预(通过蛋白质类药物、基因沉默、RNA或DNA编辑或蛋白质替代疗法的给药)的细胞内靶点。理论上讲,这样的治疗策略可用于几乎任何医学病症。具体而言,在PDIT领域内极具吸引力的候选蛋白质是CRISPR(clustered regularly interspaced short palindromic repeats(成簇规律间隔的短回文重复序列))相关蛋白,并且特别是Cas9蛋白。该最近发现的蛋白质最初是细菌蛋白质,其由细菌天然用作抗病毒机制。实际上,该蛋白质可由任意RNA序列负载,所述RNA序列具有指引蛋白质特定地至基因组中的任意基因座上的特异性。这样的基因座可能是包含突变的、缺陷基因的位点。在该位点处,则Cas9蛋白会诱导准确的剪切,即,会诱导DNA的双链断裂。然后天然存在的DNA修复机制被激活来修复所述功能失常基因位点。因此,该蛋白质能够进行高效基因编辑(特定基因序列的添加、断裂或改变)以及基因调节和修复,其可应用于整个生命树的物种。通过向细胞递送Cas9蛋白和合适的导向RNA,可将生物体的基因组在任何期望的位点上剪切,并进行编辑和修复。
如下文所示例,本发明的实施方案包括含有一个或多个“分子马达”和所述Cas9蛋白或在编辑DNA或RNA中具有作用的相关蛋白质的缀合物。本发明的另一实施方案包括治疗性蛋白质,其作为对于与特定的、生理学上重要的蛋白质的相对降低的水平相关的疾病的替代疗法给药。
根据本发明的实施方案的化合物通常包含根据不对称极性原理(如上文所解释)设计的疏水的(辛醇/水分配系数(logP>1))、偶极的、不带电荷的化学基团。如所讨论,当将分子(其为疏水的、中性的,但含有集中的局部负电荷和分散的局部正电荷)的独特结构放入膜的力场(通过其分子在磷脂膜中移动,从膜/水界面至膜中心)中,其创建矢量系统。当连接至药物,该分子会分别地将所述药物拉至膜中心。
如图1B中图示说明,根据本发明的实施方案的化合物(例如通过式IX的化合物说明)通常包含“分子马达”,所述分子马达通常是下列结构元素的组合:
(i).负极(A):通常包含至少1个选自卤素(如氟原子)和氧的负电性原子;其中在所述极包含数个负电性原子的情况下,它们以集中的、球形的(或接近球形)的排布方式排布。由于所述原子的吸电子特性和它们的空间排布,所述化合物的负极是富电子焦点。
(ii).正极(B):包含选自碳、硅、硼、磷和硫的相对正电性原子;以当被放入磷脂膜中时能够与相邻的烃链的最大化相互作用的方式排布,优选通过以线性、支化或环状链的脂族或芳族结构或者其组合的形式排布。在本发明的一个实施方案中,所述正极包含线性饱和烃链,或者类固醇基团,例如胆固醇、胆汁酸、雌二醇、雌三醇或其组合。
除了“分子马达”外,根据本发明的实施方案的化合物可包含一个或多个进一步如下所述的连接基(L)和可裂解基团(Q)。化合物可通过连接基或可裂解基团与药物(D)缀合或与之连接。
本发明的实施方案还涉及缀合至药物(如蛋白质或寡核苷酸(如siRNA或ASO))的根据本发明的实施方案的化合物用于治疗医学病症(如退行性病症、癌症、毒性或缺血性损伤、感染或免疫介导的疾病,其中特定蛋白质可在疾病病因学或发病机制中发挥作用,并且其中通过siRNA或反义机制沉默各基因的表达可在抑制疾病相关进程中具有有益效果)的用途。
例如,根据本发明的实施方案的缀合物可用作反义疗法,其为医学治疗的形式,其包括给药单链或双链核苷酸(DNA、RNA或化学类似物),所述核苷酸会结合至编码特定蛋白质的DNA,或结合至各信使RNA(mRNA)。该治疗发挥抑制基因表达并防止各蛋白质的产生的作用。作为另外的选择,本发明的缀合可包含治疗性蛋白质(如Cas9蛋白)。
在本发明的上下文中,术语“药物”(drug或medicament)涉及当向患有疾病的患者给药时,能够对所述患者发挥有益作用的化学物质。所述有益作用可以为症状的,或抵消在疾病过程中发挥作用的药剂或物质的效果。所述药物可包括给药以抑制基因表达的小分子或大分子(如蛋白质或RNA或DNA链)。例如,所述药物可包括siRNA或ASO。在一些实施方案中,药物旨在治疗退行性病症;癌症;缺血性、感染性或毒性损伤;或者免疫介导的疾病。
本发明的实施方案提供包含根据本发明的实施方案的化合物和药物的新缀合物。本发明的实施方案还提供包含所述缀合物的新药物组合物,以及基于这些药物组合物用于治疗医学病症的新方法。
根据一些实施方案,本发明的化合物和药物组合物可用于实现替代蛋白质疗法或基因疗法(例如体内siRNA或反义疗法(ASO))的更有效的性能。
根据本发明的实施方案的缀合物可在改善siRNA、ASO或治疗性蛋白质通过细胞膜或通过血脑屏障的递送中是有益的,从而改善siRNA或ASO在一个或多个方面(例如功效、毒性或药物代谢动力学)的性能。
如上所述,作为非限制性的可能的作用机制(MOA),当根据本发明的实施方案的包含药物(如siRNA或治疗性蛋白质)的缀合物位于磷脂膜内时,“分子马达”(例如如上所述)发挥将所述药物拉向膜中心的作用。理论上说,在磷脂首基的侧方运动并生成瞬间膜孔(通过其可发生药物的跨膜通过)下,接着发生膜的局部失稳。该可能的MOA在图2中图示总结。
在一个实施例中,如图2中图示呈现,缀合物包含负载物(其为siRNA、ASO或治疗性蛋白质),和用于将所述负载物捕获在细胞质中的二硫键基团。在第一阶段(A)中,由于不对称极性的原理,通过内部膜电场供能,“马达”从膜表面移动到膜中心。
在第二阶段(B)中,连接至所述“马达”的大分子集中靠近膜表面,从而扰乱水合外层。由此,发生磷脂首基的侧方运动并形成瞬间膜孔,通过其大分子被递送入细胞。所述瞬间孔的随后闭合是热力学有利的(C)。
在另一个实施方案中,siRNA在细胞质中的捕获可涵盖包括以下的方法:(i).给药Dicer底物,其包含25-30个核苷酸的双链RNA,所述RNA包含用于沉默靶基因的序列,其中所述“分子马达”连接至有义链(过客链)的3’-末端和/或连接至反义链(引导链)的5’-末端;(ii).给药根据本发明的实施方案的缀合物,其会导致药物(如siRNA)的跨膜递送和通过Dicer酶的dsRNA的裂解,从而从所述缀合物上移除所述“分子马达”,并释放所述siRNA。由于所述siRNA的大量负电荷,其会被捕获在细胞质中,并能够与RISC复合物相互作用以沉默靶基因。
在如图3所图示说明在一个实施方案中,显示利用Dicer酶将siRNA捕获在细胞质中的机制。显示Dicer底物,其包含25-30个核苷酸的双链RNA,所述RNA包含用于沉默靶基因的序列。“分子马达”(箭头)连接到siRNA的有义链(过客链)的3’-末端和/或反义链(引导链)的5’-末端,所述siRNA被设计用于沉默靶基因。在跨膜递送后,发生通过Dicer酶裂解dsRNA,将所述“分子马达”从缀合物上移除,并释放siRNA,其与RISC复合物相互作用以沉默靶基因。通过该机制,由于所述siRNA的大量负电荷,因此其被捕获在细胞质中。
根据本发明的实施方案的缀合物可由通式(I)描述:
Ey-D-Ez
(式I)
所述缀合物包括由式(I)中所示结构表示的化合物的药学上可接受的盐、水合物、溶剂合物和金属螯合物,以及所述盐的溶剂合物和水合物,其中:
D是被跨生物膜递送的药物。D可以为小分子药物、肽、蛋白质,或者天然或修饰的单链或双链DNA或RNA,如siRNA或ASO;
E表示根据本发明的实施方案的化合物;y和z为各自独立地选自0、1、2、3、4、5、6的整数;y或z中的至少一个不是0。在一个实施方案中,y=1且z=0;在另一实施方案中,y=1且z=1。E可由通式(II)描述:
(A)a-B-Q-L
(式II)
其中a为选自1、2、3或4的整数,并且A选自如下所述的式III、IV和V所示的结构;
B为饱和或部分饱和的线性、支化或环状的C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、C19、C20、C21、C22、C23、C24、C25、C26、C27、C28、C29、C30、C31、C32、C33、C34、C35、C36、C37、C38、C39、C40、C41、C42烷基、亚烷基、亚杂烷基、芳基、杂芳基;甾体基团(诸如胆固醇、胆汁酸、雌激素或其组合)的化学基团;
Q不存在或者选自酯、硫酯、酰胺、氨基甲酸酯、二硫键[-(S-S)-]、醚[-O-]、pH敏感基团和氧化还原敏感基团;
L可以不存在,或者与如上述所定义的B相同,并且可包括任选被(氧、羟基、氮、磷酸酯或硫)取代的线性、环状或支化的饱和、不饱和或部分饱和的C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、C19、C20、C21、C22、C23、C24、C25、C26、C27、C28、C29、C30、C31、C32、C33、C34、C35、C36、C37、C38、C39、C40、C41、C42烷基、亚烷基、亚杂烷基、芳基、杂芳基;甾体(诸如胆固醇、胆汁酸;雌激素)、-(O-CH2-CH2)u-结构的化学基团,其中u为1、2、3、4、5、6、7、8、9、10的整数;或其组合。
如上文所讨论,A选自如下的式III、IV、V所示的结构,其中*为与B、Q、L或D的连接点:
其中M选自-O-或-CH2-;
其中g为选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16的整数;
其中h为选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15和16的整数。
D与所述分子的基团的连接可通过醚、酯、酰胺、硫酯、硫醚或氨基甲酸酯基团。在D是寡核苷酸的情况下,可连接至核苷酸的核碱基、连接至核糖基团(如通过2’、3’或5’位),或连接至磷酸酯基团;可连接至寡核苷酸链的末端或非末端核苷酸;在D是蛋白质的情况下,其与所述分子的其它基团的连接可为通过所述蛋白质的氨基酸(如赖氨酸、谷氨酸和天冬氨酸)的侧链的连接。
在本发明上下文中,术语“寡核苷酸”可包括DNA或RNA分子,其各自是一个或多个核苷酸的单链或双链序列。各核苷酸包含含氮碱基(核碱基)、五碳糖(核糖或脱氧核糖)和磷酸酯基团。所述核碱基选自嘌呤(腺嘌呤、鸟嘌呤)和嘧啶(胸腺嘧啶、胞嘧啶、尿嘧啶)。此外,所述术语可指核苷酸的修饰形式,其中所述修饰可位于分子主链上(如硫代磷酸酯)或在核碱基中(如在RNA核糖基团的2’位的甲基化)。这些修饰可带来如寡核苷酸的改善的稳定性或改善的药物代谢动力学的性质,并且使用这样修饰的寡核苷酸也在本发明的范围内。
可连接至核苷酸的核碱基、连接至核糖基团(如通过2’、3’或5’位),或连接至磷酸酯基团。可连接至寡核苷酸链的末端或非末端核苷酸。
在一个实施方案中公开体外或体内特异性抑制基因表达的方法;所述方法包括利用本发明的缀合物,或各自的药物组合物,其中D为siRNA或ASO,其被设计为沉默编码致病蛋白质的特定基因的表达,所述致病蛋白质在疾病的病因学或发病机制中发挥作用。
因此,根据本发明的实施方案的缀合物可用于治疗医学病症。本发明的实施方案还公开用于医学治疗的方法,其包括向有需要的患者给药足够量的根据本发明的实施方案的药物组合物。在一个实施方案中,所给药的药物组合物可包含siRNA或反义寡核苷酸,其在抑制编码特定致病(即疾病相关)蛋白质的基因的表达中有活性。
在一个实施方案中,通式I的分子包含具有如式(VI)所示结构的E:
其中n和m为各自独立地选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20的整数;k为选自2、3、4、5、6、7的整数。在一个实施方案中,k=2;Z不存在或者为-S-S-,并且Q如上文所述并且为与D的连接。
在另一实施方案中,通式I的分子包含具有如式VII或VIIa所示结构的E:
在另一实施方案中,通式I的分子包含具有如式VIII所示结构的E:
其中n和m为各自独立地选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20的整数;Z不存在或者为-S-S-,并且Q(如上文所述)为与D的连接。
在另一实施方案中,通式I的分子包含具有如式IX或IXa所示结构的E:
在根据本发明的实施方案的缀合物的合成中使用的分子(也称作“前体”)也在本发明的范围内。
在一个实施方案中,这样的分子具有如式X所示的结构:
其中W为式II、III、IV、V、VI、VII、VIIa、VIII、IX或IXa中任一个的化合物。该前体可用于例如连接至寡核苷酸的5’-末端。
本发明的另一前体具有式(XI)的结构:
其中G为式II、III、IV、V、VI、VII、VIIa、VIII、IX或IXa中任一个的化合物。该前体可用于例如连接至寡核苷酸的3’-末端。
本发明的实施方案还可包括药物组合物,其包含含有I、II、III、IV、V、VI、VII、VIIa、VIII、IX或IXa中任一个的分子的缀合物和药学上可接受的盐或载体。
本发明还包括在体外或体内特异性抑制基因表达的方法。在一个实施方案中,所述方法可包括利用式I的缀合物或各自的药物组合物,其中D为siRNA或ASO(其被设计为沉默特定基因的表达,其中所述基因编码在疾病的病因学或发病机制中发挥作用的致病蛋白质);或者治疗性蛋白质。
根据本发明的实施方案的缀合物可用于治疗医学病症。本发明的实施方案包括医学治疗的方法,其包括向有需要的患者给药足够量的包含式I的缀合物的药物组合物,其中D为可用于治疗各医学病症的药物。
在一个实施方案中,所述方法用于用siRNA或ASO的基因治疗,所述方法包括向有需要的患者给药足够量的包含本发明的式I的缀合物的药物组合物,其中D为siRNA、ASO或治疗性蛋白质,其可用于抑制在特定患者的疾病中发挥作用的基因表达。
在本发明的另一实施方案中,D为被跨生物磷脂膜或通过生物屏障(如血脑屏障)递送入细胞的蛋白质。
在本发明的另一实施方案中,D为正常蛋白质,将其作为替代疗法(如替代突变的功能失常的蛋白质)给药,。
在另一实施方案中,D为在基因调节中具有作用的蛋白质,包括例如在DNA或RNA编辑(特定基因序列的添加、断裂或改变)中具有作用的蛋白质。在一个实施方案中,所述蛋白质可以为CRISPR(成簇规律间隔的短回文重复序列)相关蛋白的成员。具体而言,所述蛋白质可以为或者可包含Cas9蛋白(CRISPR相关蛋白9)、RNA引导的DNA核酸酶或其类似物。
在一个实施方案中,本发明描述用于医学病症的基因治疗的方法,所述方法包括向有需要的患者给药足够量的包含式I的缀合物(其中D为CRISPR蛋白,如Cas9)的药物组合物,将其与合适的引导寡核苷酸一起给药,由此实现所述蛋白质的递送,其通过各自的引导寡核苷酸装载入细胞,在此它们可以发挥它们的基因编辑活性。在该上下文中,引导寡核苷酸是引导Cas9蛋白到DNA上特定的基因座(位置)以修复基因物质缺陷的RNA或DNA序列。在Cas9蛋白的情况中,引导寡核苷酸是RNA。
因此,根据本发明的实施方案的缀合物以及各自的药物组合物和方法可例如对于治疗选自如癌症、毒性损伤、缺血性疾病、感染性疾病、蛋白质贮积疾病、创伤、免疫介导的疾病或退行性疾病等的医学病症是有益的。
在神经病症领域,根据本发明的实施方案的缀合物可用于例如治疗神经退行性病症,如阿尔茨海默病、运动神经元病、帕金森病、亨廷顿病、多发性硬化和克-雅二氏病(Creutzfeldt-Jacob disease)。
实施例
实施例1:合成根据本发明的实施方案的包含寡核苷酸的缀合物的一般方法:
首先,基于基因在疾病病因学或发病机制中的作用选择要沉默的基因。然后,基于本领域已知的生物信息学方法学,确定ASO的DNA序列的各siRNA的核苷酸序列(对于RISC底物通常19-21个碱基对,并且对于Dicer底物25-29个)。
合成以3'到5'的方向进行。应用固相合成,使用亚磷酰胺结构单元,其得自保护的2'-脱氧核苷(dA、dC、dG和T)、核糖核苷(A、C、G和U)或者化学修饰的核苷,如LNA(锁定核酸)或BNA(桥连核算)。结构单元顺序(以由期望的siRNA或ASO的序列要求的顺序)偶联至增长的寡核苷酸链。
在构建寡核苷酸后,将根据本发明的实施方案的化合物(E,如上述定义)作为寡核苷酸的结构单元之一加入。可将所述化合物任选地以如上所述的其前体形式加入。对于在寡核苷酸的5’-末端添加所述化合物,其前体形式的化合物可包含亚磷酰胺基团。对于在寡核苷酸的3’-末端添加化合物,其前体形式的化合物可包含乙炔或叠氮基团。通常,所述过程是完全自动的。一旦完成链的组装,产物从固相释放至溶液,脱保护并收集。然后通过高效液相色谱法(HPLC)收集并分离期望的缀合物,以高纯度地获得期望的寡核苷酸。在siRNA的情况中,将各互补RNA链分开合成,然后进行两条链的退火,以得到期望的siRNA双链RNA。
实施例2:合成根据本发明的实施方案的具体示例性化合物的方法:
根据本发明的实施方案的示例性化合物的合成从胆汁酸石胆酸(其可作为如上所述的B基团的实例)开始。
全氟叔丁醇是市售可得的,并可作为如上所述的A基团的实例。
合成如以下路线I中所述进行。在该实施例中,设计E基团连接至寡核苷酸的5’-末端,因此,在合成的最后步骤加入亚磷酰胺基团。
将25g的物质1转化为26g(定量)的甲基酯(物质2)。使26g的物质2与TBDMSCl NS 29g(87%)反应,得到NMR纯的物质3。物质3(29g)用NaBH4THF/MeOH向物质4的还原,在后处理和纯化后得到25g物质4(85%),通过NMR显示仍有少量的物质3。物质4与全氟叔丁醇的Mitsunobu反应在柱色谱法后处理和用MeOH研磨后得到33.5g(92%)物质5,之后将其脱保护,以得到甾体6。然后将甾体6(2.5g)与THP-保护的溴十四醇偶联。所述偶联进行3天,并且需要4当量的THP-保护的溴十四醇以达到完全转化。将产物通过柱色谱法纯化。在用MeOH/1,4-二氧杂环己烷(HCl,4N)/THF移除保护基(THP)后,将产物7通过柱色谱法纯化以移除杂质。获得白色固体形式的产物7(1.5g,c.y 48%)。然后将产物7通过连接亚磷酰胺基团转化为所需的化合物8。
实施例3:连接至寡核苷酸的前体和各化合物的结构
a.5’修饰:
前体:
连接至寡核苷酸:
b.3’修饰:
前体:
DMT=二甲氧三苯甲基;
CPG=定孔玻璃珠(CPG),其作为用于合成寡核苷酸的固体载体。
连接至寡核苷酸:
c.5’修饰;化合物连接到核碱基(如胸腺嘧啶):
实施例4:根据本发明的实施方案与Cas9蛋白缀合的化合物的示例性结构
缀合至Cas9蛋白的本发明的“分子马达”的结构如图4示例说明。结构基于Cas9蛋白结构的晶体学研究(Nishimazu FG等人Cell 156:935–949,2014)。根据本发明的实施方案的化合物E通过其Q基团连接至蛋白质,结合到蛋白质表面的赖氨酸侧链。为了连接,会使用活性酯。醇被转化为碳酸活性酯,其优先与氮(蛋白质赖氨酸侧链)反应而非氧(水)。反应会根据以下路线II进行:
可能的衍生化物质为:
a)光气:通过氯甲酸酯连接。
b)二琥珀酰亚胺碳酸酯(X=N-羟基琥珀酰亚胺):通过琥珀酰亚胺碳酸酯连接。
c)羰基二咪唑(CDI,X=咪唑):通过咪唑基氨基甲酸酯连接。
用任意这些基团标记蛋白质在无胺(不是Tris)的略碱性缓冲液(pH=8-9)中进行。连接点是疏水的,因此对于与蛋白质的反应需要共溶剂(通常是DMF或DMSO)。高反应性意味着更短的反应时间,但也有较低的氮/氧选择性,以及在水性缓冲液中较短的寿命。用所有活性酯,产物是氨基甲酸酯,其对酶裂解敏感。在上述三个选项中,羰基二咪唑具有最高的氮/氧选择性,以及最简单的合成。另一方面,蛋白质衍生化可花费更长时间(可能过夜)。每个蛋白质分子的E单元的数目通过预先设定期望的摩尔比率来决定。
实施例5:根据本发明的实施方案的Cy3-标记的SS 29-聚体DNA序列的体外细胞摄取和集中
细胞培养:
用GFP稳定地转染的NIH-3T3细胞系从Cell Biolabs,San-Diago,USA(cat-AKR214)获得。
所述细胞在补充有10%胎牛血清(FBS;Biological Industries cat-04-011-1A)、2mM L-谷氨酰胺(Biological Industries cat-03-020-1B)、1%Pen-Strep(Biological Industries cat-03-031-1B)和10μg/ml杀稻瘟素(Enzo cat-ALX-380-089)的Dulbecco修饰的Eagle’s培养基(DMEM;Gibco cat-41965),在37℃潮湿的含有5%CO2的培养箱中生长。
29-聚体DNA递送入细胞的评价;体外测定:
实验前一天,将对数生长期的NIH-3T3-GFP细胞以4.5x104个细胞/孔的密度用DMEM加补充剂的生长培养基(无抗生素)(500μl/孔)种于24孔板中。将各Cy3-标记的寡核苷酸在100μl/孔的Opti-Mem(Life technologies-Cat.31985062)中稀释并加入到细胞,终浓度为40-100nM。
在孵育后2、4、8和24小时评估Cy3-标记的寡核苷酸递送系统在细胞内的蓄积。孵育期后,将细胞用Hank's缓冲盐溶液(HBSS缓冲液;Biological Industries)洗涤,并进行分析。
使用Tecan200 PRO多模式读取器(激发波长548±4.5nm,发射波长580±10nm)进行Cy3阳性群的检测和定量。
将连接至Cy3-ssDNA寡核苷酸(29nt)(Syncom,the Netherlands;IDT,USA)的根据本发明的实施方案的化合物的跨膜递送与对照Cy3-ssDNA寡核苷酸(IDT,USA)比较,并表示为Cy3-阳性细胞与未处理细胞相比的百分比。
荧光显微术:
在实验前24小时,将NIH-3T3 GFP细胞以4.5x104/孔的密度接种。将生长培养基用含有连接至Cy3-ssDNA寡核苷酸(29nt)的根据本发明的实施方案的化合物(40-100nM,溶解在无DNase/RNase的水中)或对照Cy3-ssDNA(IDT,USA)的新鲜培养基替换,在37℃下孵育2、4、8和24小时。
孵育后,将细胞用HBSS洗涤并使用Olympus荧光显微镜(BX51TF;Olympus Optical,U.K.)用来自汞灯的UV照明显影。Cy3-荧光团用在470-495nm下激发和在590nm下发射来显影。GFP荧光团用在530-550nm下激发和在510-550nm下发射来显影。
在图5A和5B中描述根据本发明的实施方案的缀合物在将ss 29-聚体DNA的ASO跨脂质膜递送入细胞的生物学性能,所述ss 29-聚体DNA在其5’-末端通过磷酸酯键连接至根据本发明的实施方案的E基团(在该实施例中为式VIIa的化合物)。该构建体在5’-末端还具有iCy3-荧光团。在用GFP蛋白稳定转染的NIH-3T3细胞中评价递送。
图5A显示3T3-GFP细胞与100nM浓度的连接至Cy3-ssDNA的根据本发明的实施方案的化合物(“E-缀合物”)或Cy3-ssDNA孵育8小时时间的结果。之后,将细胞通过荧光显微镜分析(20倍放大)。
E-缀合物在上面显示,而下面是对照ASO(无根据本发明的实施方案的化合物(“E基团”))。
如所示,在对照构建体(无E基团)未表现任何显著的向细胞内递送时,E-缀合物表现摄取入几乎所有细胞。在37℃下孵育8小时时获取图像。显示与100nM的E-缀合物孵育。
通过ELISA定量分析
将3T3-GFP细胞与不同浓度(40nM和100nM)的E-缀合物或Cy3-ssDNA孵育24小时。将细胞洗涤并通过ELISA荧光计评估。将Cy3荧光水平与3T3-GFP未处理细胞比较。
如图5B中所示,在40nM和100nM的剂量下,与对照相比,缀合物表现出4倍和3倍增加的向细胞内的递送。
实施例6:在体外通过siRNA敲减mRNA
评价25-27bp dsRNA在稳定表达GFP(绿色荧光蛋白)报告基因的细胞中的抑制活性。为此目的,将表达GFP的稳定转染的NIH 3T3细胞用25-27聚体dsRNA双链体(各自为10-40nM)转染。为获得GFP基因敲减的持续时间的定量评估,会进行时程实验,观察转染后的GFP表达。
实验前一天,将对数生长期的NIH-3T3-GFP细胞以4.5x104细胞/孔的密度用DMEM加补充剂的生长培养基(无抗生素)(500μl/孔)种于24孔板中。将各Cy3-标记的寡核苷酸在100μl/孔的Opti-Mem(Life technologies-Cat.31985062)中稀释,并加入到细胞,终浓度为40-100nM。
在孵育后24、48、72小时评价细胞中GFP的mRNA敲减。孵育期后用Hank's缓冲盐溶液(HBSS缓冲液;Biological Industries)洗涤细胞并进行分析。
使用Tecan200 PRO多模式读取器(激发波长488±4.5nm和发射波长535±10nm)进行GFP阳性群的检测和定量。
将连接至25-27bp dsRNA(Syncom,the Netherlands;IDT,USA)的根据本发明的实施方案的化合物的敲减活性与对照25-27bp dsRNA(IDT,USA)比较,并表达为GFP阳性细胞与未处理细胞相比的百分比。
实施例7:siRNA细胞内捕获的机制,包括给药Dicer底物
在本发明的实施方案中,在细胞质内捕获siRNA的方法是基于Dicer酶的活性,在处理双链RNA中,将其以19-21个碱基对的大小剪切,其适合与RISC复合物相互作用以用于基因沉默。所述方法包括:(i).给药Dicer底物,其包含25-30个核苷酸的双链RNA,所述RNA包含用于沉默靶基因的序列,其中根据本发明的实施方案的化合物连接至有义链(过客链)的3’-末端和/或连接至反义链(引导链)的5’-末端;(ii).siRNA的跨膜递送;以及(iii).通过Dicer酶裂解dsRNA,由此释放siRNA,以与RISC复合物相互作用,用于沉默靶基因。
对于体外Dicer裂解测定法,可将siRNA双链体(100pmol)在20ml的20mM Tris pH 8.0、200mM NaCl、2.5mM MgCl2与1单位的重组人Dicer(Stratagene)孵育24小时。在15%非变性聚丙烯酰胺凝胶中分离各反应物的3mL等分部分(15pmol RNA),用GelStar(Ambrex)染色,并使用紫外激发显影。使用Oligo HTCS系统(Novatia)(其由ThermoFinniganTSQ7000、Xcalibur数据系统、ProMass数据处理软件和Paradigm MS4HPLC(Michrom BioResources)组成)进行用Dicer处理之前和之后的双链体RNA的电喷雾离子化液相色谱质谱法(ESILCMS)。
- 还没有人留言评论。精彩留言会获得点赞!
- 能够与配体偶联的单甲基缬氨酸化合物的制造方法与工艺
- 烟梗纤维微波降解制备烟用料液方法及应用与流程
- 利用特异性挥发物筛选昆虫相关嗅觉蛋白的方法与制造工艺
- 小分子共价抑制剂的计算机筛选方法及其在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂的应用与制造工艺
- 一种合成抗原的分子量检测方法及应用与制造工艺
- 一种用于MALDI‑TOF‑MS分析小分子的氧化锌沸石复合材料及其制备方法与制造工艺
- 用于原位收集根系挥发物的试验装置及方法与制造工艺
- 一种核壳型纳米香精胶囊的制备方法与制造工艺
- 小分子共价抑制剂在制备抑制S-腺苷甲硫氨酸脱羧酶的药物上的应用及其筛选方法与制造工艺
- Linderolide H在制备治疗鼻炎药物中的应用的制造方法与工艺