用于治疗肿瘤的抑制性寡核苷酸的制作方法
本申请要求2014年2月7日提交的美国临时专利申请系列号61/937,376(其完整内容在此通过引用整体并入)的权益和优先权。
发明领域
本发明涉及用于治疗肿瘤诸如B细胞淋巴瘤的寡核苷酸及其用途。
发明背景
免疫系统保护人体免受细菌、寄生虫、真菌、病毒感染和防止肿瘤细胞的生长。免疫可被分类为先天免疫和适应性免疫。先天免疫应答通常在感染后立即发生,以对感染性疾病提供早期屏障,而适应性免疫应答较晚发生,产生抗原特异性长期保护性免疫。
然而,免疫应答有时可能是不希望的并造成免疫介导的病症,包括自身免疫性疾病、移植排斥、超敏反应、与微生物对宿主的免疫系统的过度刺激相关的疾病以及Toll样受体(TLR)介导的疾病。Toll样受体(TLR)介导的疾病是由Toll样受体(TLR)的激活引起的病症。
TLR是识别微生物来源的分子结构(病原体相关分子模式或PAMP)的受体家族。表达TLR的免疫细胞在结合PAMP后被活化。TLR受体识别一系列病原体来源的产物并被活化。细菌的脂多糖(LPS)被TLR4识别,脂磷壁酸和二酰化脂肽被TLR2-TLR6二聚体识别,三酰化脂肽被TLR2-TLR1二聚体识别,合成的或来源于病毒或细菌的含CpG寡核苷酸(CpG ODN)被TLR9识别,细菌鞭毛蛋白被TLR5识别,酵母聚糖被TLR2-TLR6二聚体识别,来自呼吸道合胞病毒(RSV)的F蛋白被TLR4识别,病毒来源的双链RNA(dsRNA)和聚I:C(dsRNA的合成类似物)被TLR3识别;病毒DNA被TLR9识别,单链病毒RNA(VSV和流感病毒)和合成的鸟苷类似物诸如咪唑并喹啉和咪喹莫特被TLR7和TLR8识别(Foo Y.Liew,等Nature Reviews Immunology.第5卷,June 2005,446-458)。导致I型IFN的诱导的信号传导机制取决于激活的TLR而不同。它们涉及干扰素调节因子IRF,其是已知在抗病毒防御、细胞生长和免疫调控中发挥关键作用的转录因子家族。3种IRF(IRF3、IRF5和IRF7)用作病毒介导的TLR信号传导的直接转导器。TLR3和TLR4活化IRF3和IRF7(Doyle S.等Immunity.2002 17(3):251-63),而TLR7和TLR8活化IRF5和IRF7(Schoenemeyer A.等J Biol Chem.2005 280(17):17005-12)。此外,已显示通过TLR9 1配体CpG-A刺激的I型IFN产生由磷酸肌醇3-激酶(PI3K)和mTOR介导(Costa-Mattioli M等Nat Rev Drug Discov,20109:293-307)。
发明概述
在一个方面,本发明提供了用于治疗受试者的B细胞淋巴瘤的方法,所述受试者已被诊断为患有特征在于MYD88中的突变的B细胞淋巴瘤并且需要这样的治疗,所述方法包括:向所述受试者施用包含治疗有效量的具有5’-(CCT)n-3’(其中n是2至50的整数)的序列的寡核苷酸和药学上可接受的载体的药物组合物。
在一些实施方案中,所述寡核苷酸具有5’-(CCT)nCm-3的序列,n为6至16的整数,m是0、1或2。
在一些实施方案中,B细胞淋巴瘤选自巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型以及胃粘膜相关淋巴样组织(MALT)淋巴瘤。
在一些实施方案中,MYD88中的突变包括L265P、M232T、S243N、或T294P。
在一些实施方案中,其中所述寡核苷酸包含选自以下序列的序列:
5'-cctcctcctcctcctcctcctcctc-3’(SEQ ID NO:1),
5'-cctcctcctcctcctcctcctcctcc-3’(SEQ ID NO:2),
5'-cctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:3),
5'-cctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO:4),
5'-cctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO:5),
5'-cctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:6),
5'-cctcctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO:7),
5'-cctcctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO:8),
5'-cctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:9),
5'-cctcctcctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO:10),
5'-cctcctcctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO:11),
5'-cctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:12),
5'-cctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:13),
5'-cctcctcctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO:14),
5'-cctcctcctcctcctcct-3’(SEQ ID NO:15),和
5'-cctcctcctcctcctcctcct-3’(SEQ ID NO:16)。
在一些实施方案中,寡核苷酸的磷酸骨架是未经修饰的。
在一些实施方案中,寡核苷酸的磷酸骨架是部分或完全硫代磷酸酯修饰的。
在一些实施方案中,寡核苷酸包含化学修饰。
在一些实施方案中,寡核苷酸在5’-(CCT)n-3’的序列的每一个末端还包含一个或多个核苷酸。
在一些实施方案中,通过口服、肠内、肠胃外或局部施用的途径或通过吸入施用寡核苷酸。
在一些实施方案中,将寡核苷酸与Btk抑制剂、PI3Kδ抑制剂、IRAK抑制剂、抗-CD20单克隆抗体、SYK抑制剂或Bcl-2抑制剂组合施用。
通过引用并入
本说明书中提及的所有出版物、专利和专利申请通过引用并入本文,其程度如同每个单独的出版物、专利或专利申请被具体地和单独地指出通过引用并入。
附图简述
本发明的新颖特征具体地示于所附权利要求中。本发明的特征和优势的更好理解将通过参考示出举例说明性实施方案的以下详细描述获得,其中利用了本发明的原理,并且其附图:
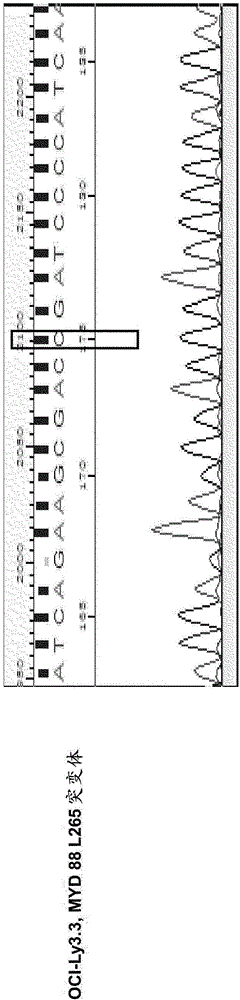
图1描绘了确认OCI-Ly3.3的MYD88 L265P突变体的存在(图1,右图),以及OCI-Ly19不具有该突变体(图1,左图)。与先前的报道一致,OCI-Ly3.3具有纯合的MYD88 L265P突变体。
图2描绘了0.3uM和1uM的3种TLR7/9拮抗剂(称为(CCT)8、(CCT)12和(CCT)12M)导致对OCI-Ly3.3细胞的细胞生长抑制但不导致对OCI-Ly19细胞的细胞生长抑制。
图3描绘了所有3种TLR7/9拮抗剂能够抑制IL-10从OCI-Ly3.3细胞分泌。
发明详述
在下文中参考用于举例说明的示例性应用描述本发明的几个方面。应当理解的是,许多具体的细节、关系和方法被示出来提供对本发明的充分理解。然而,相关领域的普通技术人员将容易地认识到本发明可以在没有一个或多个具体细节的情况下实施或利用其它方法来实施。本发明不受行为或事件的示例的顺序限制,因为一些行为可以以与其它行为或事件不同的顺序进行和/或与其它行为或事件同时进行。
此外,并非需要所有示例的行为或事件来执行根据本发明的方法学。
本文所使用的术语仅仅是为了描述具体实施方案的目的,并非旨在限制本发明。如本文中所用,除非上下文明确地另有所指,否则单数形式“一个/种(a)”,“一个/种(an)”和“该(the)”意欲也包括复数形式。此外,就术语“包括”、“含有”、“具有”,“拥有”、“有”或其变型用于发明详述和/或权利要求中而言,这样的术语旨在是以与术语“包含”类似的方式的包含性的。
术语“约”或“大致”意指在如由本领域技术人员测定的特定值的可接受误差范围内,其将部分取决于该值是如何被测量或测定的,即测量系统的限制。例如,“约”可意指按照每本领域的实践,在1个或多于1个标准偏差内。或者,“约”可意指给定值的多至20%,优选多至10%,更优选多至5%,更优选多至1%的范围。或者,特别地对于生物系统或过程,该术语可意指在值的量级内,优选在5倍内,更优选在2倍内。当在本申请和权利要求中描述特定值时,除非另有所指,否则应当假定术语“约”意指在特定值的可接受的误差范围内。
I.定义和缩写
除非另有定义,否则本文中使用的所有技术和科学术语一般具有与本发明所属的领域内的普通技术人员通常理解的含义相同的含义。一般地,本文中使用的术语以及细胞培养、分子遗传学、有机化学和核酸化学以及杂交中的实验室方法是本领域中公知的和常用的那些。标准技术用于核酸和肽合成。技术和方法通常按照本领域和各种一般参考文献中的常规方法执行,其提供在整个本文件中。本文中使用的术语以及下文中所述的分析化学和有机化学中的实验室方法在是本领域中公知的和常用的术语和方法。标准技术或其改进用于化学合成和化学分析。
II.组合物
本发明提供了用于治疗受试者的癌症诸如B细胞淋巴瘤的方法和组合物,所述受试者已被诊断为患有B细胞淋巴瘤(优选地特征在于MYD88中的突变)并且需要这样的治疗,所述治疗通过向受试者施用包含治疗有效量的能够抑制TLR9的寡核苷酸的药物组合物来进行。
本发明的寡核苷酸强烈地抑制TLR9活化。含CpG寡核苷酸(CpG ODN)已知为TLR9激动剂[D.M.Klinman,Nat.Rev.,Immunol.4(2004)249–258]。
本文中的“寡核苷酸”意指多个核苷酸,即包含连接于磷酸基团和可交换的有机碱基的糖(例如脱氧核糖)的分子,所述有机碱基是取代的嘧啶(Py)(例如,胞嘧啶(C)、胸腺嘧啶(T))或取代的嘌呤(Pu)(例如,腺嘌呤(A)或鸟嘌呤(G))。如本文中所用,术语寡核苷酸是指寡脱氧核糖核苷酸(ODN)。寡核苷酸可从现有的核酸来源(例如,基因组或cDNA)获得,但优选是合成的。本发明的寡核苷酸可通过多种可在市场上获得的自动化核酸合成仪合成。这些寡核苷酸被称为合成的寡核苷酸。
据记载,TLR9激动剂激活先天免疫和适应性免疫应答(Arthur M.Krieg.Nature Reviews Drug Discovery,第5卷June 2006,471-484)。含CpG寡核苷酸(CpG ODN)是TLR9激动剂[D.M.Klinman,Nat.Rev.,Immunol.4(2004)249–258]。基于功能特性,CpG ODN被分为3种类型(Tomoki Ito,等Blood,2006,第107卷,Num 6:2423-2431)。A型CpG ODN活化人类浆细胞样树突细胞(pDC)以产生大量I型干扰素(IFN-α/β)以及强烈地活化天然杀伤细胞(NK细胞)。B型CpG ODN主要活化B细胞,导致其增殖和抗体分泌。C型CpG ODN具有A型和B型CpG ODN的活性。作为TLR9激动剂,CpG ODN诸如CpG2216或CpG2006或CpG2395可被内吞进入细胞区室,在所述区室中它们被暴露于TLR9并激活TLR9。在pDC中,TLR9活化起始特征在于促炎细胞因子[IL-6、肿瘤坏死因子α(TNFα)]的分泌、I型干扰素(IFN)的分泌和IFN诱导的趋化因子的分泌的快速先天免疫应答。通过IFN依赖性和IFN非依赖性途径,先天免疫细胞包括天然杀伤(NK)细胞、单核细胞和嗜中性粒细胞被pDC二次活化。通过TLR9活化的B细胞具有大大增强的对抗原刺激的敏感性并且高效地分化成抗体分泌细胞,并因此促成适应性免疫应答,尤其是体液免疫应答。通过TLR9活化的pDC分泌IFNα,所述IFNα驱动pDC至淋巴结和其它二级淋巴组织的迁移和群集,在所述淋巴结和其它二级淋巴组织中pDC活化幼稚和记忆T细胞,帮助可溶性蛋白抗原至CD8+细胞毒性T淋巴细胞(CTL)的交叉呈递,并促进强的TH1偏向性细胞CD4和CD8T细胞应答。基于上述发现,很明显的是,拮抗CpG ODN的活性的试剂可用于通过抑制先天和适应性免疫应答来治疗或预防免疫介导的病症。
一般地,方法包括向受试者施用包含治疗有效量的寡核苷酸和任选地药学上可接受的载体的药物组合物,所述寡核苷酸为例如具有5’-(CCT)n-3’的序列的寡核苷酸,其中n是2至50的整数(包括2和50)。
在一些实施方案中,寡核苷酸在5’-(CCT)n-3’的序列的每一个末端还包含一个或多个核苷酸。
在一些实施方案中,寡核苷酸具有5’-(CCT)nCm-3的序列,n为6至16的整数,并且m为0、1或2。
在一些实施方案中,寡核苷酸包含选自以下序列的序列:
5'-cctcctcctcctcctcctcctcctc-3’(SEQ ID NO:1),
5'-cctcctcctcctcctcctcctcctcc-3’(SEQ ID NO.:2),
5'-cctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:3),
5'-cctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO.:4),
5'-cctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO.:5),
5'-cctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:6),
5'-cctcctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO.:7),
5'-cctcctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO.:8),
5'-cctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:9),
5'-cctcctcctcctcctcctcctcctcctcctcctc-3’(SEQ ID NO.:10),
5'-cctcctcctcctcctcctcctcctcctcctcctcc-3’(SEQ ID NO.:11),
5'-cctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:12),
5'-cctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:13),
5'-cctcctcctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’(SEQ ID NO.:14),
5'-cctcctcctcctcctcct-3’(SEQ ID NO.:15)和
5'-cctcctcctcctcctcctcct-3’(SEQ ID NO.:16)。在这些ODN中,碱基是未经修饰的或经化学修饰,诸如是硫代磷酸酯修饰的。
在一些实施方案中,本发明的寡核苷酸不包含化学修饰。
在一些实施方案中,本发明的寡核苷酸包含化学修饰。
本发明中公开的寡核苷酸相较于天然DNA可包括各种化学修饰,牵涉包括磷酸二酯核苷间桥、核糖单位和/或天然核苷碱基(腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶)。所述修饰可在寡核苷酸的合成过程中或在合成之后发生。在合成过程中,经修饰的碱基可被掺入在内部或掺入在其末端上。在合成后,可使用活性基团进行修饰(通过氨基改性剂,通过3'或5'羟基,或通过磷酸基团)。
根据本发明的寡核苷酸相较于由天然DNA组成的相同序列的寡核苷酸,可具有一个或多个修饰,其中每一个修饰位于特定的磷酸二酯核苷间桥上和/或位于特定核糖单位上和/或位于特定天然核苷碱基位置上。化学修饰包括本发明的寡核苷酸的“骨架修饰”。如本文中所用,本发明的寡核苷酸的经修饰的骨架包括,但不限于“硫代磷酸骨架”,其是指核酸分子的稳定化的糖磷酸骨架,其中在至少一个核苷酸间键联上非桥连磷酸氧被硫替代。
在一些实施方案中,在每一个核苷酸间键联上非桥连磷酸氧被硫替代。其它骨架修饰表示利用非离子型DNA类似物的修饰,所述非离子型DNA类似物为例如烷基-和芳基-膦酸酯(其中带电荷的膦酸酯氧被烷基或芳基取代)、磷酸二酯和烷基磷酸三酯(其中带电荷的氧部分是烷基化的)。
在一些实施方案中,寡核苷酸的磷酸骨架是未经修饰的。
在一些实施方案中,寡核苷酸的磷酸骨架是部分或完全硫代磷酸酯修饰的。
在一些实施方案中,寡核苷酸包含选自以下序列的序列:
5’-CCtCCTCCTCCTCCTCCTCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:21),
5’-CCtCCtCCTCCTCCTCCTCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:22),
5’-CCtCCtCCtCCTCCTCCTCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:23),
5’-CCtCCtCCtCCtCCTCCTCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:24),
5’-CCtCCtCCtCCtCCCCTCtCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:25),
5’-CCtCCtCCtCCtCCtCCtCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:26),
5’-CCtCCtCCtCCtCCtCCtCCtCCTCCTCCTCCTCCT-3’(SEQ ID NO.:27),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCTCCTCCTCCT-3’(SEQ ID NO.:28),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCTCCTCCTCCT-3’(SEQ ID NO.:29),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCtCCTCCTCCT-3’(SEQ ID NO.:30),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCtCCtTCCTCCT-3’(SEQ ID NO.:31),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCtCCtCCtCCT-3’(SEQ ID NO.:32),
5’-CCtCCtCCtCCtCCtCCtCCtCCtCCtCCtCCtCCt-3’(SEQ ID NO.:33),
5’–CCtCCTCCTCCTCCTCCtCCTCCTCCTCCTCCTCCT-3’(SEQ ID NO.:34),
5’-CCTCCtCCTCCTCCTCCTCCTCCtCCTCCTCCTCCT-3’(SEQ ID NO.:35),
5’-CCTCCTCCtCCTCCTCCTCCTCCTCCtCCTCCTCCT-3’(SEQ ID NO.:36),
5’-CCTCCTCCTCCTCCtCCTCCTCCTCCTCCTCCtCCT-3’(SEQ ID NO.:37),
5’–CCtCCTCCTCCTCCtCCTCCTCCTCCtCCTCCTCCT-3’(SEQ ID NO.:38),
5’–CCTCCtCCTCCTCCTCCtCCTCCTCCTCCtCCTCCT-3’(SEQ ID NO.:39),
5’–CCTCCTCCtCCTCCTCCTCCtCCTCCTCCTCCtCCT-3’(SEQ ID NO.:40),
5’–CCTCCTCCTCCtCCTCCTCCTCCtCCTCCTCCTCCt-3’(SEQ ID NO.:41),
5’-CCtCCTCCTCCtCCTCCTCCtCCTCCTCCtCCTCCT-3’(SEQ ID NO.:42),
5’-CCTCCtCCTCCTCCtCCTCCTCCtCCTCCTCCtCCT-3’(SEQ ID NO.:43),
5’-CCTCCTCCtCCTCCTCCtCCTCCTCCtCCTCCTCCt-3’(SEQ ID NO.:44),
5’-CCtCCTCCtCCTCCtCCTCCtCCTCCtCCTCCTCCT-3’(SEQ ID NO.:45),
5’-CCTCCTCCtCCTCCtCCTCCtCCTCCtCCtCCTCCT-3’(SEQ ID NO.:46),
5’-CCTCCTCCTCCTCCtCCTCCtCCTCCtCCtCCtCCT-3’(SEQ ID NO.:47),
5’-CCTCCTCCTCCTCCTCCTCCtCCTCCtCCtCCtCCt-3’(SEQ ID NO.:48),
5’-CCtCCTCCtCCTCCtCCTCCtCCTCCtCCTCCtCCT-3’(SEQ ID NO.:49),
5’-CCTCCtCCTCCtCCTCCtCCTCCtCCTCCtCCTCCt-3’(SEQ ID NO.:50),
其中对于SEQ ID NO.21-50,大写字母表示该碱基是硫代磷酸酯修饰的,小写字母表示该碱基是未经修饰的。
在一些实施方案中,寡核苷酸是硫代磷酸酯/磷酸二酯嵌合体。
化学修饰还包括本发明中公开的寡核苷酸的碱基取代。取代的嘌呤和嘧定可以是C-5丙炔嘧啶和7-脱氮-7-取代的嘌呤。取代的嘌呤和嘧啶包括但不限于腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶以及其它天然和非天然存在的核碱基。本发明的寡核苷酸的化学修饰还包括寡核苷酸的碱基的修饰。经修饰的碱基是在化学上与通常在DNA中发现的天然存在的碱基诸如T、C、G和A不同但与这些天然存在的碱基共有基本化学结构的任何碱基。
在一些实施方案中,本发明的寡核苷酸通过使用胞苷衍生物来修饰。术语“胞苷衍生物”是指胞苷样核苷酸(不包括胞苷),术语“胸苷衍生物”是指胸苷样核苷酸(不包括胸苷)。另外,本发明的寡核苷酸可通过在寡核苷酸的任一或两个末端上连接二醇诸如四乙二醇或六乙二醇来进行化学修饰。
除了在Py-Pu二核苷酸上的膦酰乙酸酯或膦酰乙酸酯样键联外,寡核苷酸可具有另外的骨架修饰。稳定化的核苷酸间键联是相较于磷酸二酯核苷酸间键联对体内降解(例如,通过外切或内切核酸酶)相对耐受的核苷酸间键联。除了膦酰乙酸酯和膦酰乙酸酯样键联以外,寡核苷酸还可含有其它稳定化的核苷酸间键联,包括,但不限于硫代磷酸酯、二硫代磷酸酯、甲基膦酸酯和甲基硫代磷酸酯。其它稳定化的核苷酸间键联包括但不限于:肽、烷基和脱磷酸。与其它稳定化的键联一样,膦酰乙酸酯核苷酸间键联具有减小的对核酸酶降解的易感性和增强的激活RNA酶H的能力。因此,例如磷酸二酯寡核苷酸而非膦酰乙酸酯寡核苷酸对核酸酶消化易感,然而磷酸二酯寡核苷酸和膦酰乙酸酯寡核苷酸均活化RNA酶H。在一些实施方案中,Py-Pu寡核苷酸包括至少一个磷酸二酯核苷酸间键联。除了在优选的内部位置上的膦酰乙酸酯或膦酰乙酸酯样核苷酸间键联以外,寡核苷酸还可包含抗降解的5'和3'末端。这样的抗降解的末端可包括相对于对应的未修饰末端导致增强的针对外切核酸酶消化的抗性的任何合适的修饰。例如,5’和3’末端可通过在其中包含骨架的至少一个磷酸修饰来稳定化。在一个实施方案中,每一个末端上骨架的至少一个磷酸修饰独立地是硫代磷酸酯、二硫代磷酸酯、膦酰乙酸酯、膦酰乙酸酯样、甲基膦酸酯或甲基硫代磷酸酯核苷酸间键联。在另一个实施方案中,抗降解的末端包括在3’末端通过肽或酰胺键联连接的一个或多个核苷酸单位。
术语“核酸”和“寡核苷酸”还包括诸如在碱基和/或糖中具有取代或修饰的核酸或寡核苷酸。例如,它们包括具有共价附接至低分子量有机基团而非2'位置上的羟基且非5'位置上的磷酸基团或羟基的骨架糖的核酸。因此,经修饰的核酸可包括2'-O-烷基化的脱氧核糖基团。另外,经修饰的核酸可包括糖诸如阿拉伯糖或2'-氟阿拉伯糖替代脱氧核糖。因此,核酸在骨架组成上可以是异质的,从而含有连接在一起的聚合物单位的任何可能的组合诸如肽-核酸(其具有拥有核酸碱基的氨基酸骨架)。在本发明的说明书中,寡核苷酸不是反义寡核苷酸、核酶或适体。
核酸还包括取代的嘌呤和嘧啶诸如C-5丙炔嘧啶和7-脱氮-7-取代的嘌呤修饰的碱基(Wagner RW等,(1996)Nat Biotechnol 14:840-4)。嘌呤和嘧啶包括但不限于腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶、5-甲基胞嘧啶、5-羟基胞嘧啶、5-氟胞嘧啶、2-氨基嘌呤、2-氨基-6-氯嘌呤、2,6-二氨基嘌呤、次黄嘌呤以及其它天然和非天然存在的核碱基,取代的和未取代的芳香族部分。其它这样的修饰对于本领域技术人员来说是公知的。
寡核苷酸可以是DNA或RNA。在一个实施方案中,本发明的寡核苷酸是包含核糖和脱氧核糖的混合骨架的DNA/RNA杂合分子。DNA/RNA杂合寡核苷酸经常表现出增强的活性。在一个实施方案中,这些DNA/RNA杂合寡核苷酸是单链的。在另一个实施方案中,全部或部分的寡核苷酸是双链的。在一个实施方案中,本发明的寡核苷酸是具有一级和二级结构的共价闭合的哑铃形分子的形式。在一个实施方案中,这样的环状寡核糖核苷酸包括通过间插双链区段连接的两个单链环。在一个实施方案中,至少一个单链环包括本发明的免疫刺激性DNA基序。本发明的其它共价闭合的哑铃形分子包括嵌合DNA/RNA分子,其中例如,双链区段至少部分是DNA(例如,同二聚体dsDNA或异二聚体DNA:RNA)并且至少一个单链环包括本发明的免疫刺激性DNA基序。或者,嵌合分子的双链区段是DNA。
本发明的寡核苷酸还可以包括其它修饰。这些修饰包括非离子型DNA类似物,诸如烷基-和芳基-磷酸酯(其中带电荷的膦酸酯氧被烷基或芳基取代),磷酸二酯和烷基磷酸三酯(其中带电荷的氧部分是烷基化的)。还已显示在任一或两个末端上含有二醇诸如四乙二醇或六乙二醇的核酸显著抗核酸酶降解。
本发明的寡核苷酸相较于天然RNA和DNA可包括各种化学修饰和取代,牵涉磷酸二酯核苷酸间桥、β-D-核糖单位和/或天然核苷酸碱基(腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶、尿嘧啶)。化学修饰的实例对于本领域技术人员是已知的,并描述于,例如,Uhlmann,E.等,(1990)Chem Rev 90:543;"Protocols for Oligonucleotides and Analogs"Synthesis and Properties&Synthesis and Analytical Techniques,S.Agrawal,编辑,Humana Press,Totowa,USA 1993;Crooke,ST.等,(1996)Annu Rev Pharmacol Toxicol 36:107-129;和Hunziker,J.等,(1995)Mod Synth Methods 7:331-417中。相较于由天然DNA或RNA组成的相同序列的寡核苷酸,根据本发明的寡核苷酸可以具有一个或多个修饰,其中每一个修饰位于特定的磷酸二酯核苷酸间桥和/或位于特定的β-D-核糖单位和/或位于特定的天然核苷酸碱基位置。
例如,本发明涉及这样的寡核苷酸,其可以包括一个或多个修饰,并且其中每一个修饰独立地选自:a)经修饰的核苷酸间桥对位于核苷酸的3’和/或5’末端的磷酸二酯核苷酸间桥的替代,b)脱磷酸桥对位于核苷酸的3'和/或5'末端的磷酸二酯桥的替代,c)另一个单位对来自糖磷酸骨架的糖磷酸单位的替代,d)经修饰的糖单位对β-D-核糖单位的替代,以及e)经修饰的核苷酸碱基对天然核苷酸碱基的替代。
位于核苷酸的3'和/或5'末端的磷酸二酯核苷酸间桥可被经修饰的核苷酸间桥替代,其中所述经修饰的核苷酸间桥例如选自硫代磷酸酯、二硫代磷酸酯、NR1R2-氨基磷酸酯,硼烷磷酸酯、α-羟基苄基膦酸酯、磷酸-(CrC2i)-O-烷基酯、磷酸-[(C6-Ci2)芳基-(Ci-C2i)-O-烷基酯、(d-CeJ烷基膦酸酯和/或(C6-C12)芳基膦酸酯桥,(C7-C12)-α-羟甲基-芳基(例如,于WO 95/01363中公开的),其中(C6-Ci2)芳基、(Cβ-C2o)芳基和(C6-Ci4)芳基任选地被卤素、烷基、烷氧基、硝基、氰基取代,并且其中R1和R2彼此独立地为氢、(CrCi8)-烷基、(C6-C2o)-芳基、(C6-Ci4)-芳基-(CrC8)-烷基,优选卤素、(CrC8)-烷基,优选(Ci-C4)-烷基和/或甲氧基乙基,或R1和R2与携带它们的氮原子一起形成可额外地含有选自O、S和N的另外的杂原子的5-6元杂环。
脱磷酸桥对位于核苷酸的3'和/或5’末端的磷酸二酯桥的替代(脱磷酸桥描述于例如Uhlmann E和Peyman A in"Methods in Molecular Biology,"第20卷,"Protocols for Oligonucleotides and Analogs,"S.Agrawal,编辑,Humana Press,Totowa 1993,第16章,第355ff页中),其中脱磷酸桥例如选自脱磷酸桥甲缩醛(formacetal)、3'-硫代甲缩醛(3'-thioformacetal)、甲基羟胺、肟、亚甲基二甲基-亚肼基、二亚甲基砜和/或甲硅烷基。来自糖磷酸骨架(即,糖磷酸骨架由糖磷酸酯单位组成)的糖磷酸酯单位(即,一起形成糖磷酸酯单位的β-D-核糖和磷酸二酯核苷酸间桥)可被另一种单位替代,其中所述另一种单位例如适合构建“吗啉代-衍生物”寡聚物(如例如Stirchak EP等,(1989)Nucleic Acids Res 17:6129-41中描述的),即,例如,被吗啉代-衍生物单位替代;或构建聚酰胺核酸("PNA";如例如Nielsen PE等,(1994)Bioconjug Chem 5:3-7中描述的),即,例如,被PNA骨架单位替代,例如被2-氨基乙基甘氨酸替代。
β-D-核糖单位或β-D-2'-脱氧核糖单位可被经修饰的糖单位替代,其中所述经修饰的糖单位例如选自α-D-2'-脱氧核糖、α-L-2'-脱氧核糖、β-L-2'-脱氧核糖、β-L-核糖、2'-F-21-脱氧核糖、2'-F-2'-脱氧-阿拉伯糖、2'-O-(Ci-C6)烷基-核糖,优选地2'-O-(CrC6)烷基-核糖是2'-O-甲基核糖、2I-O-(C2-C6)烯基-核糖、21-[O-(Ci-C6)烷基-O-(Ci-C6)烷基]-核糖、21-NH2-21-脱氧核糖、β-D-木-呋喃糖、α-阿拉伯呋喃糖、2,4-二脱氧-β-D-赤-已-吡喃糖和碳环(描述于例如Froehler,J.(1992)Am Chem Soc 114:8320中)和/或开链糖类似物(描述于例如Vandendriessche等,(1993)Tetrahedron 49:7223中)和/或双环糖类似物(描述于例如Tarkov,M.等,(1993)HeIv Chim Acta 76:481中)。
在一些实施方案中,糖是2'-O-甲基核糖、2'-脱氧核糖、2'-氟-2'-脱氧核糖、2'-氨基-2'脱氧核糖、2'-O-烷基-核糖或3'-O-烷基-核糖和/或2'-O-4'-C-烯烃基核糖,诸如2'-O-4'-C-亚甲基核糖(也称为LNA)。
核酸还包括取代的嘌呤和嘧啶诸如C-5丙炔嘧啶和7-脱氮-7-取代的嘌呤修饰的碱基(Wagner,R.W.等,(1996)Nat Biotechnol 14:840-4)。嘌呤和嘧啶包括但不限于腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶,以及基它天然和非天然存在的核碱基,取代和未取代的芳香部分。
经修饰的碱基是在化学上与通常在DNA和RNA中发现的天然存在的碱基诸如T、C、G、A和U不同但与这些天然存在的碱基共有基本化学结构的任何碱基。经修饰的核苷酸碱基可以例如选自次黄嘌呤、尿嘧啶、二氢尿嘧啶、假尿嘧啶、2-硫代尿嘧啶、4-硫代尿嘧啶、5-氨基尿嘧啶、5-(Ci-C6)-烷基尿嘧啶、5-(C2-Cβ)-烯基尿嘧啶、5-(C2-C6)-炔基尿嘧啶、5-(羟甲基)尿嘧啶、5-氯尿嘧啶、5-氟尿嘧啶、5-溴尿嘧啶、5-碘-尿嘧啶、2.4-二氟-甲苯和3-硝基吡咯、5-羟基胞嘧啶、5-(CrC6)-烷基胞嘧啶、5-(C2-C6)-烯基胞嘧啶、5-(C2-Ce)-炔基胞嘧啶、5-氯胞嘧啶、5-氟胞嘧啶、5-溴胞嘧啶、N2-二甲基鸟嘌呤、2,4-二氨基-嘌呤、8-氮杂嘌呤、取代的7-脱氮嘌呤,优选地7-脱氮-7-取代的和/或7-脱氮-8-取代的嘌呤、5-羟甲基胞嘧啶、N4-烷基胞嘧啶,例如,N4-乙基胞嘧啶、5-羟基脱氧胞苷、5-羟甲基脱氧胞苷、N4-烷基脱氧胞苷,例如,N4-乙基脱氧胞苷、6-硫代脱氧鸟苷,和硝基吡咯的脱氧核糖核苷酸、C5-丙炔基嘧啶,和二氨基嘌呤例如2,6-二氨基嘌呤、肌苷、5-甲基胞嘧啶、2-氨基嘌呤、2-氨基-6-氯嘌呤、次黄嘌呤或天然核苷酸碱基的其它修饰。该列表意欲是举例性的,并且不被解释为是限制性的。
在本文中,“Py”用于指嘧啶,并且在一些实施方案中,指含有胞嘧啶或经修饰的胞嘧啶的核苷酸。如本文中所用,经修饰的胞嘧啶是胞嘧啶的天然存在或非天然存在的嘧啶碱基类似物,其可替代该碱基而不削弱寡核苷酸的免疫刺激活性。经修饰的胞嘧啶包括但不限于5-取代的胞嘧啶(例如,5-甲基-胞嘧啶、5-氟-胞嘧啶、5-氯-胞嘧啶、5-溴胞嘧啶、5-碘-胞嘧啶、5-羟基-胞嘧啶、5-羟甲基-胞嘧啶、5-二氟甲基-胞嘧啶,和未取代的或取代的5-炔基-胞嘧啶)、6-取代的胞嘧啶、N4-取代的胞嘧啶(例如,N4-乙基-胞嘧啶)、5-氮杂-胞嘧啶、2-巯基-胞嘧啶、异胞嘧啶、假-异胞嘧啶、具有稠环系统的胞嘧啶类似物(例如,N.N'-丙烯胞嘧啶或吩噁嗪)和尿嘧啶及其衍生物(例如,5-氟-尿嘧啶、5-溴-尿嘧啶、5-溴乙烯基-尿嘧啶、4-硫代-尿嘧啶、5-羟基-尿嘧啶、5-丙炔基-尿嘧啶)。一些优选的胞嘧啶包括5-甲基胞嘧啶、5-氟-胞嘧啶、5-羟基-胞嘧啶、5-羟甲基-胞嘧啶和N4-乙基-胞嘧啶。在本发明的另一个实施方案中,胞嘧啶碱基被通用碱基(例如,3-硝基吡咯、P-碱基)、芳环系统(例如,氟苯或二氟苯)或氢原子(dSpacer)取代。
在本文中,“Pu”用于指嘌呤或经修饰的嘌呤。在一些实施方案中,Pu是鸟嘌呤或经修饰的鸟嘌呤碱基。如本文中所用,经修饰的鸟嘌呤是鸟嘌呤的天然存在的或非天然存在的嘌呤碱基类似物,其可替代该碱基而不削弱寡核苷酸的免疫刺激活性。经修饰的鸟嘌呤包括但不限于7-脱氮鸟嘌呤、7-脱氮-7-取代的鸟嘌呤(诸如7-脱氮-7-(C2-C6)炔基鸟嘌呤)、7-脱氮-8-取代的鸟嘌呤、次黄嘌呤、N2-取代的鸟嘌呤(例如,N2-甲基-鸟嘌呤)、5-氨基-3-甲基-3H,6H-噻唑并[4,5-d]嘧啶-2,7-二酮、2,6-二氨基嘌呤、2-氨基嘌呤、嘌呤、吲哚、腺嘌呤、取代的腺嘌呤(例如,N6-甲基-腺嘌呤、8-羟基腺嘌呤)、8-取代的鸟嘌呤(例如,8-羟基鸟嘌呤和8-溴鸟嘌呤)和6-硫鸟嘌呤。在本发明的另一个实施方案中,鸟嘌呤碱基被通用碱基(例如,4-甲基吲哚、5-硝基-吲哚和K-碱基)、芳香环系统(例如,苯并咪唑或二氯-苯并咪唑、1-甲基-1H-[1,2,4]三唑-3-羧酸酰胺)或氢原子(dSpacer)取代。
本发明还包括具有不常见的核苷酸间键联(包括5'-5'、2'-2'、2'-3'和2'-5'核苷酸间键联)的寡核苷酸。在本发明的一些方面,寡核苷酸具有一个或多个可及的5'末端是有利的。有可能产生具有两个这样的5'末端的经修饰的寡核苷酸。这可例如通过将两个寡核苷酸通过3'-3'键联连接以产生具有一个或两个可及的5'末端的寡核苷酸来实现。3'-3'键联可以是磷酸二酯、硫代磷酸酯、膦酰乙酸酯或任何其它经修饰的核苷酸间桥。用于实现这样的键联的方法在本领域中是已知的。例如,这样的键联已描述于Seliger,H.等Oligonucleotide analogs with terminal 3'-3'-and 5'-5'-internucleotidic linkages as antisense inhibitors of viral gene expression,Nucleotides&Nucleotides(1991),10(1-3),469-77和Jiang等,Pseudo-cyclic oligonucleotides:in vitro and in vivo properties,Bioorganic&Medicinal Chemistry(1999),7(12),2727-2735中。在一个实施方案中,从免疫刺激性DNA基序排除这样的不常见键联,即使一个或多个这样的键联可存在于聚合物内的其它地方。对于具有游离末端的聚合物,一个3'-3'核苷酸间键联的包含可导致具有两个游离5'末端的聚合物。相反地,对于具有游离末端的聚合物,一个5'-5'核苷酸间键联的包含可导致具有两个游离3'末端的聚合物。另外,其中键联不是磷酸二酯、硫代磷酸酯、膦酰乙酰酯或其它经修饰的桥的3'3'-、5-5'-、2'-2'、2'-3'-和2'-5'-连接的核酸可使用附加的间隔子,诸如三-或四-乙二醇磷酸部分来制备(Durand,M.等,Triple-helix formation by an oligonucleotide containing one(dA)12and two(dT)12sequences bridged by two hexaethylene glycol chains,Biochemistry(1992),31(38),9197-204,美国专利No.5658738和美国专利No.5668265)。或者,非核苷酸接头可从乙二醇、丙二醇衍生,或使用标准亚磷酰胺化学从无碱基脱氧核糖(dSpacer)单位衍生(Fontanel,Marie Laurence等,Sterical recognition by T4 polynucleotide kinase of non-nucleosidic moieties 51-attached to oligonucleotides;Nucleic Acids Research(1994),22(11),2022-7)。非核苷酸接头可被掺入一次或多次,或与彼此组合,从而允许两个待连接的ODN的3'端之间的任何期望的距离。
寡核苷酸可含有二倍器(doubler)或三倍器(trebler)单元(Glen Research,Sterling,VA),特别是具有3'-3'键联的那些经修饰的寡脱氧核糖核苷酸类似物。在一个实施方案中,二倍器单元可基于1,3-双-[5-(4,4'-二甲氧基三苯甲基氧)戊基酰胺基]丙基-2-[(2-氰乙基)-(N,N-二异丙基)]-亚磷酰胺。在一个实施方案中,三倍器单元可以基于Tris-2,2,2-[3-(4,4'-二甲氧基三苯甲基氧)丙氧甲基]乙基-[(2-氰乙基)-(N,N-二异丙基)]-亚磷酰胺的掺入。通过多个二倍器、三倍器或其它倍增器单元对经修饰的寡核糖核苷酸的分枝导致为本发明的进一步实施方案的树状大分子。分枝的经修饰的寡核糖核苷酸类似物相较于该类似物的非分枝形式,可导致具有不同免疫作用的受体(特别是针对免疫刺激性RNA和DNA的组合的受体)诸如TLR3、TLR7、TLR8和TLR9的交联。另外,分枝的或其它方式的多聚体类似物的合成可稳定DNA免受降解,并且可使弱的或部分有效的DNA序列能够产生治疗上有用的水平的免疫活性。经修饰的寡脱氧核糖核苷酸类似物还可包含从肽修饰剂或寡核苷酸修饰剂(Glen Research)产生的接头单元。此外,经修饰的寡脱氧核糖核苷酸类似物可含有一个或多个通过肽(酰胺)键联连接至聚合物的天然或非天然氨基酸残基。
在一些实施方案中,可通过连接一个或多个聚乙二醇(PEG)链来修饰寡核苷酸。PEG化的寡核苷酸可通过减少肾清除来在体内延长循环时间。
3'-5'、5'-5'、3'-3\2'-2'、2'-3'和2'-5'核苷酸间键联可以是直接的或间接的。本上下文中的直接键联是指如本文所公开的无间插接头部分的磷酸酯或经修饰的磷酸酯键联。间插接头部分是与如本文中公开的磷酸酯或经修饰的磷酸酯键联不同的有机部分,其可包括,例如,聚乙二醇、三乙二醇、六乙二醇、dSpacer(即,无碱基脱氧核苷酸)、二倍器单元或三倍器单元。键联优选由C、H、N、O、S、B、P和卤素组成,含有3至300个原子。具有3个原子的实例是连接例如一个核苷酸的3'-羟基与第二寡核苷酸的3'-羟基的缩醛键联(ODN1-3'-O-CH2-O-3'-ODN2)。具有约300原子的实例是PEG-40(聚乙二醇40)。优选键联是磷酸二酯、硫代磷酸酯、甲基膦酸酯、氨基磷酸酯、硼烷膦酸酯、酰胺、醚、硫醚、缩醛、硫缩醛、尿素、硫脲、磺酰胺、Schiff碱和二硫键联。还可能使用Solulink BioConjugation系统。
如果寡核苷酸由两个或更多个序列部分组成,则这些部分可以相同或不同。因此,在具有3'3'-键联的寡核苷酸中,序列可以是相同的5'-ODN1-3'3'-ODN1-5'或不同的5'-ODN1-3'3'-ODN2-5'。此外,各种寡核苷酸部分以及连接它们的接头的化学修饰可以是不同的。因为短的寡核苷酸的摄取似乎不如长寡核苷酸的摄取高效,两个或更多个短的序列的连接导致增强的免疫刺激。短的寡核苷酸的长度优选是2-20个核苷酸,更优选3-16个核苷酸,但最优选5-10个核苷酸。优选的是具有两个或更多个未连接的5'-末端的连接的寡核苷酸。
寡核苷酸的部分序列还可通过非核苷酸接头来连接。如本文中所用,“非核苷酸接头”是指不是核苷酸或其聚合物(即,多核苷酸)的任何接头元件(其中核苷酸包含嘌呤或嘧啶核碱基和糖磷酸酯),特别是无碱基接头(dSpacers)、三乙二醇单位或六乙二醇单位。进一步优选的接头是烷氨基接头,诸如C3、C6、C12氨基接头,以及还有烷基硫醇接头,诸如C3或C6硫醇接头。寡核苷酸还可通过可被烷基或取代的烷基进一步取代的芳香族残基连接。
其它稳定化的寡核苷酸包括:非离子型DNA类似物,诸如烷基-和芳基-磷酸酯(其中带电荷的膦酸酯氧被烷基或芳基替代)、磷酸二酯和烷基磷酸三酯(其中带电荷的氧部分被烷基化)。还已显示在任一或两个末端含有二醇诸如四乙二醇或六乙二醇的核酸显著抗核酸酶降解。
III.特征在于MYD88的L265P突变的B细胞淋巴瘤
在一个方面,本发明提供了用于治疗受试者的癌症诸如B细胞淋巴瘤的方法和组合物,所述受试者已诊断为患有优选地特征在于MYD88中的突变的B细胞淋巴瘤并且需要这样的治疗。
如本文所用,术语“受试者”是指动物。优选地,动物是哺乳动物。受试者还指例如灵长类动物(例如,人)、牛、绵羊、山羊、马、狗、猫、兔、大鼠、小鼠、鱼、鸟等。在优选的实施方案中,受试者是人。
某些遗传限定的B细胞淋巴瘤是几种类型的B细胞淋巴瘤中在MYD88中具有致癌突变的那些B细胞淋巴瘤,包括,但不限于,巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型和胃粘膜相关淋巴样组织(MALT)淋巴瘤。
本文中的“激动剂”意指结合细胞的受体并诱导应答的物质。激动剂通常模拟天然存在的物质诸如配体的作用。
本文中的“拮抗剂”意指衰减激动剂的作用的物质。
本发明的寡核苷酸强烈地抑制受CpG ODN刺激的细胞因子,这表明本发明的寡核苷酸可用作用于治疗与TLR9活化相关的疾病诸如B细胞淋巴瘤,特别是某些遗传限定的在MYD88中具有致癌突变的B细胞淋巴瘤(包括巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型以及胃粘膜相关淋巴样组织(MALT)淋巴瘤)的补救。
TLR已牵涉在骨髓分化初级应答基因88(MYD88)中具有致癌突变的B细胞淋巴瘤的发病机制(Lim KH等2013AACR Abs.)。MYD88是TLR和白细胞介素1受体(IL-1R)信号传导的衔接分子。在TLR或IL-1R刺激后,MYD88被招募至活化的受体复合物作为同二聚体,其与IL-1R相关激酶(IRAK)4复合以激活IRAK1(Fitzgerald KA等Nature.2001 413 78–83)。活化的IRAK1与肿瘤坏死因子受体相关因子6(TRAF6)结合,其催化多聚泛素至TRAF6上的添加。泛素的添加活化TAK/TAB复合物,其进而磷酸化干扰素调节因子(IRF),从而导致核因子κB(NF-κB)释放并运输至细胞核。细胞核中的NF-κB诱导促炎基因表达。最近,已在几种类型的B细胞淋巴瘤中鉴定了MYD88中的致癌突变(Ngo V等Nature.2011 115-119;Treon SP等NEJM.2012 826-833;Poulain S等Blood.2013 4504-4511)。在WM(91%)、ABC DLBCL(30%)和MALT淋巴瘤(9%)中已以较高的频率观察到MYD88中的体细胞L265P突变的存在。还在MYD88中观察到另外的突变,其以较低的频率存在于ABC和生发中心B细胞样(GCB)DLBCL亚型中(Ngo V等Nature.2011 470:115-119)。
据报道,表达MYD88 L265P的ABC DLBCL细胞的存活通过该突变体得到维持(Yang G等2013Blood 1222-1232)。L265P突变体通过自发组装含有IRAK1和IRAK4的蛋白质复合体,从而导致IRAK4激酶活性、IRAK1磷酸化、NF-κB信号传导、STAT3的JAK激酶活化以及IL-6、IL-10和干扰素-β的分泌来促进ABC DLBCL细胞的存活(Ngo V等Nature.2011 470:115-119)。这些研究表明L265P是功能获得性驱动者突变以及异常的MYD88信号传导途径是WM和ABC DLBCL的发病机制所不可或缺的。
已显示MYD88 L265P突变衔接TLR7与TLR9以赋予ABC DLBCL细胞存活益处(Ngo V等Nature.2011 470:115-119)。MyD88 L265P癌蛋白组成性地结合TLR7和TLR9,由此放大从这些受体发出的信号。TLR7或TLR9的敲低强有力地抑制了ABC DLBCL细胞系中的NF-κB活性,并促进细胞凋亡。对于TLR7和TLR9运输和信号传导所必需的已知的蛋白(包括UNC93B1、PRAT4A或CD14)的耗尽对于ABC DLBCL系是致命的。使用组织蛋白酶抑制剂zFA-fmk或羟氯喹对溶酶体区室中的TLR7和TLR9功能的药物抑制减弱了MyD88信号传导和降低了ABC DLBCL系的存活率(Lin KH等Cancer Research 2013第73卷(8)Abstract 2332)。在配体结合中具有缺陷的TLR7或TLR9突变体在于ABC DLBCL系中敲低内源性TLR7或TLR9表达后,不能促进ABC DLBCL系的存活(Poulain S等Blood 2013 4504-4511)。
抑制性ODN对TLR7和TLR9的抑制增加了具有MYD88 L265P突变的肿瘤细胞的细胞死亡率,抑制细胞因子产生和信号传导途径的关键组分(Liang XQ等Blood 2010 5041-5052;Zhang YS等Inter Immunophamarcology.2012 446-453)。在表达MYD88 L265P的WM细胞系中,MYD88/IRAK信号传导的抑制减弱了NF-κB信号传导和肿瘤细胞的存活。相应地,WM细胞存活通过MYD88 L265P过表达得以增强(Treon SP等NEJM.2012 826-833;Ansell SM等Blood.2012 2699;Poulain S等Blood.2013 4504-11)。
在一些实施方案中,B细胞淋巴瘤选自巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型以及胃粘膜相关淋巴样组织(MALT)淋巴瘤。
在一些实施方案中,MYD88中的突变包括L265P、M232T、S243N或T294P。
已证明特定TLR的抑制是治疗某些遗传限定的B细胞淋巴瘤的有用方法(Lim KH等,2013AACR Abs.)。在这些研究中,已在MYD88(介导TLR和白细胞介素1受体(IL-1R)信号传导的衔接蛋白)中鉴定了特定的体细胞L265P突变。已显示所述突变衔接TLR7与TLR9以赋予肿瘤细胞存活益处,而TLR7和TLR9的抑制导致具有该突变的肿瘤细胞的细胞死亡率增加。在91%的巨球蛋白血症(WM)、30%的弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型和9%的胃粘膜相关淋巴样组织(MALT)淋巴瘤中观察到以较高的频率存在的MYD88中的L265P突变。在MYD88中还观察到另外的突变,其以较低的频率存在于ABC和生发中心B细胞样(GCB)DLBCL亚型中。
本发明的寡核苷酸强烈抑制细胞因子产生和信号传导途径的关键组分,增加了携带MYD88中的L265P突变的人类淋巴瘤细胞系的细胞死亡率。本发明的寡核苷酸可用作用于治疗具有MYD88中的致癌突变的某些遗传限定的B细胞淋巴瘤的补救。所述B细胞淋巴瘤包括,但不限于,巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)的活化B细胞样(ABC)亚型以及胃粘膜相关淋巴样组织(MALT)淋巴瘤。
本发明提供了用于使用本领域中已知的和/或本文中提供的方法筛查具有MYD88突变的癌症患者,并利用本文中提供的寡核苷酸治疗这样的患者的方法和组合物。
可在基因组DNA水平、mRNA水平或蛋白质水平上检测MYD88基因。使用本领域中已知的方法获得需要测试的受试者的生物样品。任选地处理所述生物样品以获得蛋白质、RNA和/或DNA,所述蛋白质、RNA和/或DNA进而被用于测定以检测MYD88突变。
A.生物样品
本文中的“生物样品”意指怀疑含有MYD88突变多核苷酸或多肽或其片段的任何生物样品,并且可以包括细胞,从细胞分离的染色体(例如,伸展的中期染色体),基因组DNA(在溶液中或结合于固体支持物诸如用于Southern分析),RNA(在溶液中或结合于固体支持物诸如用于Northern分析),cDNA(在溶液中或结合于固体支持物),来自细胞、血液、尿液、骨髓、或组织等的提取物。
可从其中存在或正在发展特征在于MYD88的突变的癌症的任何哺乳动物获得用于本发明的方法的实践的生物样品。在一个实施方案中,哺乳动物是人。人候选者可以是目前正在利用寡核苷酸诸如本文提供的寡核苷酸治疗的,或考虑用所述寡核苷酸治疗的患者。在另一个实施方案中,哺乳动物是大型动物,诸如马或牛,而在其它实施方案中,哺乳动物是小型动物,诸如狗或猫,已知所有这些动物发生癌症,包括肺癌。
来自哺乳动物癌症的包含细胞(或细胞的提取物)的任何生物样品适用于本发明的方法。还可使用如所描述的肿瘤标志物、细胞角蛋白标志物或其它阴性选择方法(参见Ma等,Anticancer Res.23(1A):49-62(2003))从血清获得循环肿瘤细胞。血清和骨髓样品对于白血病患者可以是特别优选的。对于涉及实体瘤诸如肉瘤和癌的癌症,生物样品可包含获得自肿瘤活检的细胞,其可能按照标准临床技术来获得。
循环肿瘤细胞(“CTC”)可以,例如,使用在Vita-AssaysTM、Vita-CapTM和商标下出售的试剂盒和试剂(可从Vitatex,LLC(Johnson and Johnson公司)商购获得)来进行纯化。描述了用于分离CTC的其它方法(参见,例如,PCT公开号WO/2002/020825,Cristofanilli等,New Engl.J.of Med.351(8):781-791(2004)和Adams等,J.Amer.Chem.Soc.130(27):8633-8641(2008年7月))。在特定的实施方案中,可分离循环肿瘤细胞(“CTC”)并将其鉴定为源自肺。
B.MYD88突变多肽的检测
在一些实施方案中,MYD88突变(例如MYD88 L265P)通过免疫测定进行检测。MYD88 L265P蛋白质或肽被生成来产生特异于MYD88 L265P蛋白的抗体(单克隆或多克隆的)。随后将这样的抗体用于测定以检测MYD88 L265P的存在。
通常使用Rspo融合特异性试剂来检测MYD88 L265P。本文中的“MYD88 L265P特异性试剂”意指能够特异性结合、检测和/或定量生物样品中的表达的MYD88 L265P多肽的存在/水平的任何生物或化学试剂。该术语包括,但不限于,下文中论述的优选抗体和试剂,并且等效试剂在本发明的范围之内。
适用于本发明的方法的实践的试剂包括MYD88 L265P多肽特异性抗体。本发明的MYD88 L265P特异性抗体是特异性结合本发明的MYD88 L265P多肽(例如对应于本文所提供的MYD88 L265P序列的肽),但基本上不结合野生型MYD88的分离的抗体。
人MYD88 L265P特异性抗体也可结合于其它哺乳动物物种例如鼠或兔中的高度同源和等同的表位肽序列,反之亦然。用于实施本发明的方法的抗体包括(a)特异性结合靶多肽的单克隆抗体,(b)特异性结合靶多肽的纯化的多克隆抗体,(c)如上述(a)-(b)中描述的结合其它非人物种(例如小鼠、大鼠)中的等同的和高度同源的表位或磷酸化位点的抗体,和(d)上述(a)-(c)的片段,其结合被本文公开的示例性抗体结合的抗原(或更优选地表位)。
本文中的“抗体”或“多个抗体”意指所有类型的免疫球蛋白,包括IgG、IgM、IgA、IgD和IgE。抗体可以是单克隆或多克隆的,并且可以是任何来源的物种,包括(例如)小鼠、大鼠、兔、马或人,或可以是嵌合抗体。参见,例如,M.Walker等,Molec.Immunol.26:403-11(1989);Morrision等,Proc.Nat'l.Acad.Sci.81:6851(1984);Neuberger等,Nature 312:604(1984))。抗体可以是根据美国专利号4,474,893(Reading)或美国专利号4,816,567(Cabilly等)中公开的方法产生的重组单克隆抗体。抗体也可以是按照美国专利号4,676,980(Segel等)中公开的方法产生的化学构建的特异性抗体。
本发明并不限定于抗体的用途,而是包括以融合蛋白或截短蛋白特异性方式结合与用于本发明的方法的MYD88 L265P特异性抗体所结合的表位基本上相同的表位的等效分子,诸如蛋白质结合结构域或核酸适体。参见,例如,Neuberger等,Nature 312:604(1984)。这样的等效非抗体试剂可适合用于在下文中进一步描述的本发明的方法。
可按照标准技术,通过用抗原(包含期望的融合蛋白特异性表位(例如本文中描述的Rspo融合蛋白的融合接合处))免疫合适的动物(例如,兔、山羊等),从该动物收集免疫血清,和从所述免疫血清分离多克隆抗体,以及按照已知的方法纯化具有所需特异性的多克隆抗体,来产生在实施本发明的方法中有用的多克隆抗体。抗原可以是包含所需表位序列的合成肽抗原,并按照公知的技术选择和构建。参见,例如,ANTIBODIES:A LABORATORY MANUAL,第5章,第75-76页,Harlow&Lane编辑,Cold Spring Harbor Laboratory(1988);Czernik,Methods In Enzymology,201:264-283(1991);Merrifield,J.Am.Chem.Soc.85:21-49(1962)).可如下文中进一步所述地筛选和分离如本文中所述产生的多克隆抗体。
单克隆抗体还可有益地用于本发明的方法,并可按照Kohler和Milstein.Nature 265:495-97(1975);Kohler和Milstein,Eur.J.Immunol.6:511(1976)(也参见,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,Ausubel等编辑(1989))中的公知的技术在杂交瘤细胞系中产生。所产生的单克隆抗体是高度特异的,并且提高了由本发明提供的测定方法的选择性和特异性。例如,可将含有适当的抗原(例如含有Rspo-PTPRK或Rspo-EIF3E融合多肽的融合接合处的合成肽)的溶液注射入小鼠,并在足够的时间之后(按照常规技术),处死小鼠并获取脾细胞。随后通常在聚乙二醇存在的情况下,通过将所述脾细胞与骨髓瘤细胞融合来永生化它们,以产生杂交瘤细胞。可以例如如1997年10月7日发布的美国专利号5,675,063(K.Knight)所述产生兔融合杂交瘤。随后将杂交瘤细胞生长在合适的选择培养基诸如次黄嘌呤-氨基蝶呤-胸苷(HAT)中,并如下文所述筛选上清液的具有所需特异性的单克隆抗体。可通过常规方法,诸如沉淀、离子交换或亲和层析等从组织培养上清液回收分泌的抗体。
还可通过本领域技术人员已知的重组技术在大肠杆菌(Escherichia coli)中产生单克隆Fab片段。参见,例如,W.Huse,Science 246:1275-81(1989);Mullinax等,Proc.Nat'l Acad.Sci.87:8095(1990)。如果一个同种型的单克隆抗体对于特定应用是优选的,则可通过从初始融合物选择来直接制备特定同种型,或通过使用分离类别转换变体的同胞选择技术(Steplewski,等,Proc.Nat'l.Acad.Sci.,82:8653(1985);Spira等,J.Immunol.Methods,74:307(1984))从分泌不同同种型的单克隆抗体的亲代杂交瘤选择来二次制备特定同种型。可利用PCR克隆单克隆抗体的抗原组合位点,并在大肠杆菌中将单链抗体产生为噬菌体展示的重组抗体或可溶性抗体(参见,例如ANTIBODY ENGINEERING PROTOCOLS,1995,Humana Press,Sudhir Paul editor.)。
更进一步地,美国专利号5,194,392,Geysen(1990)描述了检测或测定作为与目标抗体的特定互补位(抗原结合位点)互补的表位的拓扑等同物(即,“模拟表位”)的单体的序列(氨基酸或其它化合物)的一般方法。更一般地,本方法涉及检测或测定单体的序列,所述单体是与特定的目标受体的配体结合位点互补的配体的拓扑等同物。类似地,美国专利号5,480,971,Houghten等(1996)公开了线性C1-C-烷基全烷基化寡肽和这样的肽的组和文库,以及使用这样的寡肽组和文库测定优先结合目标受体分子的全烷基化寡肽的序列的方法。因此,本发明的具有表位的肽的非肽类似物也可通过这些方法来常规制造。
可按照标准技术就表位和融合蛋白特异性筛选可用于本发明的方法的抗体(无论是多克隆还是单克隆的)。参见,例如Czernik等,Methods in Enzymology,201:264-283(1991)。例如,可通过ELISA针对肽文库筛选抗体,以确保对于所需抗原的特异性以及,如果需要的话,确保只与例如本发明的MYD88 L265P多肽而非与野生型Rspo或野生型MYD88的反应性。还可通过免疫印迹针对含有靶蛋白的细胞制剂来测试抗体,以确认只与所需靶的反应性和确保没有明显的与野生型MYD88的结合。融合蛋白特异性抗体的产生、筛选及使用对于本领域技术人员来说是已知的,并已被描述。参见,例如,美国专利公开号20050214301,Wetzel等,2005年9月29日。
用于本发明的方法的MYD88 L265P特异性抗体可表现出一些有限的与其它融合蛋白中的类似融合表位或与野生型MYD88中的表位的交叉反应性。这并不意外,因为大多数抗体表现出一定程度的交叉反应性,并且抗肽抗体往往与对免疫肽具有高同源性或同一性的表位交叉反应。参见,例如,Czernik,同上。与其它融合蛋白的交叉反应可容易地通过免疫印迹连同具有已知分子量的标志物来表征。可检查交叉反应性蛋白的氨基酸序列,以鉴定与抗体所结合的MYD88 L265P多肽序列高度同源或同一的位点。可在肽柱上使用抗体纯化,通过负向选择除去不期望的交叉反应性(例如选择出结合任一野生型MYD88的抗体)。
可用于实施本文所公开的方法的本发明的MYD88 L265P特异性抗体理想地特异于人融合多肽,但不局限于只结合人物种本身。本发明包括也结合其它哺乳动物物种(例如:小鼠、大鼠、猴)中的保守以及高度同源或同一的表位的抗体的产生和使用。其它物种中的高度同源或同一的序列可通过与人MYD88 L265P的标准序列比较,诸如使用BLAST,来容易地鉴定。
本发明的方法中使用的抗体可通过例如流式细胞术(FC)、免疫组织化学(IHC)和/或免疫细胞化学(ICC)来进一步表征,以及通过所述技术来进一步验证用于特定的测定形式中使用。还可有利地将抗体缀合于荧光染料(例如Alexa488,PE)或标签诸如量子点,连同其它信号传导(磷酸-AKT、磷酸-Erk 1/2)和/或细胞标志物(细胞角蛋白)抗体,用于多参数分析。
C.MYD88 L265P多核苷酸的检测
由本发明提供的MYD88 L265P特异性试剂还包括适合用于MYD88 L265P多核苷酸的检测的核酸探针和引物。在本文中描述了这样的探针在测定诸如聚合酶链反应(PCR)扩增中的特殊用途。
在一些实施方案中,通过PCR,如常规PCR、实时PCR(Q-PCR)或数字PCR(包括微滴数字PCR)检测MYD88 L265P。将一对引物用于扩增融合基因。基于待扩增的融合基因序列设计引物。优选地,一个引物与Rspo基因的第一序列杂交,第二引物与融合伴侣基因的第二序列杂交。可在可如本领域中已知的进行优化的条件下,对cDNA(如使用生物样品从RNA制备的)或基因组DNA进行PCR。
应当理解的是,可将检测本发明的MYD88 L265P多核苷酸的所有方法(例如,PCR)与检测MYD88 L265P多核苷酸或MYD88 L265P多肽的其它方法组合。例如,可在检测生物样品的遗传物质中的(例如,循环肿瘤细胞中的)MYD88 L265P多核苷酸后,进行样品的蛋白质的免疫印迹分析或免疫组织化学(IHC)分析以确定MYD88 L265P多核苷酸在生物样品中是否实际上被表达为MYD88 L265P多肽。这样的免疫印迹或IHC分析可使用特异性结合于由检测的MYD88 L265P多核苷酸编码的多肽的抗体来进行。
在一些实施方案中,使用其中将定制的融合基因微阵列用于检测来自癌症样本的MYD88 L265P的微阵列,通过杂交来检测MYD88 L265P基因。寡核苷酸被设计来使得能够组合嵌合转录物接合处的测量与单个融合伴侣的逐外显子测量。参见Skotheim,RI;Thomassen,GO;Eken,M;Lind,GE;Micci,F;Ribeiro,FR;Cerveira,N;Teixeira,MR等A universal assay for detection of oncogenic fusion transcripts by oligo microarray analysis.Molecular Cancer 8:5.(2009)。
在一些实施方案中,使用分支DNA测定,通过杂交检测MYD88 L265P突变。在这些实施方案中,将定制的杂交和信号扩增测定诸如分支DNA测定用于检测来自癌症样本的裂解溶液中的Rspo融合转录物。捕获延伸器探针和标签延伸器探针的序列来源于MYD88 L265P基因的外显子序列。
在一些实施方案中,通过测序诸如Sanger测序或下一代测序检测MYD88 L265P突变。
可使用多种方法进行通过延伸测序引物或通过延伸延伸的产物进行的测序。例如,可利用标记的可逆终止子或通过利用标记的寡核苷酸的连接进行测序。可使用任何商购可得的方法,诸如可从公司如Illumina,Inc.(San Diego,CA)和Life Technologies(Ion Torrent)商购获得的基于可逆终止子的测序方法进行测序。
在一些实施方案中,使用克隆单分子阵列(Solexa,Inc/Illumina,Inc.)或利用可逆终止子化学的合成测序(SBS)进行高通量测序。这些技术部分描述于例如,美国专利号6,969,488;6,897,023;6,833,246;6,787,308和美国公开申请号20040106130;20030064398;20030022207;和Constans,A.,The Scientist 2003,17(13):36中。
IV.药物制剂和施用
本发明还涉及包含本发明的化合物或其药学上可接受的盐以及一种或多种药学上可接受的载体的药物组合物或药物制剂。
在本文中,“药物组合物”意指包含治疗有效量的本发明的寡核苷酸,具有或不具有药学上可接受的载体的组合物。所述药物组合物可包含本发明的一种或多种寡核苷酸。所述组合物包括但不限于水溶液或盐水溶液、颗粒、气雾剂、丸剂、颗粒剂、粉剂、片剂、包衣片剂、(微)胶囊、栓剂、糖浆剂、乳剂、混悬剂、乳膏、滴剂和适合用于多种药物递送系统的其它药物组合物。可肠胃外、口服、经直肠、阴道内、腹膜内、局部(以如粉剂、软膏、凝胶、滴剂或透皮贴剂的剂型)、经颊或作为口或鼻喷雾施用所述组合物。在所有情况下,所述组合物在制造和储存的条件下必须是无菌的和稳定的,并且经防腐处理以防止微生物污染。用于胃肠外注射的本发明的药物组合物包含药学上可接受的无菌水性或非水性溶液、分散体、悬浮液或乳液,以及用于在使用前立即重建成无菌可注射液或分散体的无菌粉剂。可将本发明的寡核苷酸悬浮在水性载体中,例如,悬浮在pH约3.0至约8.0,优选pH约3.5至约7.4、3.5至6.0或3.5至约5.0的等渗缓冲液中。缓冲溶液包括柠檬酸钠-柠檬酸,和磷酸钠-磷酸,以及乙酸钠-乙酸缓冲液。对于口服施用,将用可食用载体配制所述组合物以形成粉末片剂、丸剂、锭剂、胶囊、液体、凝胶、糖浆、浆液、悬浮液等。对于固体组合物,常规无毒固体载体可包括药物级的甘露糖醇、乳糖、淀粉或硬脂酸镁。对于口腔施用,所述组合物将是常规方式的片剂或锭剂。对于吸入,所述组合物将是来自加压包装或喷雾器的气溶胶喷雾剂或干粉,并且可由本领域技术人员来选择。在某些情况下,为了延长本发明的寡核苷酸的作用,还通过持续释放系统来适当地施用本发明的寡核苷酸。可将本发明的寡核苷酸在具有差的水溶性的晶体或无定型材料的液体悬浮液中使用,以减缓寡核苷酸的释放。或者,通过将寡核苷酸溶解或悬浮在疏水材料(诸如可接受的油媒介物)中来实现寡核苷酸的胃肠外施用的药物形式的延迟释放。通过将寡核苷酸捕获在脂质体或微乳剂或其它生物可降解的半渗性聚合物材料诸如聚交酯-聚乙交酯、聚(原酸酯)和聚(酸酐)中来产生可注射贮库形式。
可使用多种施用途径或模式将本文所述的包括药学上可接受的载体诸如加成盐或其水化物的化合物递送至患者。
如本文所用,术语“药学上可接受的载体/赋形剂”包括任何和所有溶剂、分散介质、包衣、表面活性剂、抗氧化剂、防腐剂(例如,抗菌剂、抗真菌剂)、等渗剂、吸收延迟剂、盐、药物、药物稳定剂、粘合剂、赋形剂、崩解剂、润滑剂、甜味剂、调味剂、染料、诸如这样的的物质及其组合,这对于本领域普通技术人员来说是已知的(参见,例如,Remington's Pharmaceutical Sciences,第18版Mack Printing Company,1990,pp.1289-1329,通过引用并入本文)。除了任何常规载体与活性成分不相容外,设想了其在治疗或药物组合物中的使用。
如本文中所用,术语“药物可接受的盐”是指保留本发明的化合物的生物学有效性和性质,并且不是在生物学上或其它方面不期望的盐。在许多情况下,本发明的化合物能够凭借氨基和/或羧基或与其类似的基团(例如,酚或羟肟酸)的存在形成酸和/或碱盐。药学上可接受的酸加成盐可用无机酸和有机酸来形成。可从其衍生盐的无机酸可包括,例如,盐酸、氢溴酸、硫酸、硝酸、磷酸等。可从其衍生盐的有机酸包括,例如,乙酸、丙酸、乙醇酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸等。药学上可接受的碱加成盐可用无机和有机碱形成。可从其衍生盐的无机碱包括,例如,钠、钾、锂、铵、钙、镁、铁、锌、铜、锰、铝等;特别优选的是铵盐、钾盐、钠盐、钙盐和镁盐。可从其衍生盐的有机碱包括,例如,伯胺、仲胺和叔胺、取代的胺,包括天然存在的取代的胺、环胺、碱性离子交换树脂等,具体地诸如异丙胺、三甲胺、二乙胺、三乙胺、三丙胺和乙醇胺。本发明的药学上可接受的盐可通过常规化学方法从母体化合物、碱性或酸性部分合成。一般地,这样的盐可通过将这些化合物的游离酸形式与化学计量的量的合适的碱(诸如Na、Ca、Mg或K氢氧化物、碳酸盐,碳酸氢盐等)反应,或通过将这些化合物的游离碱形式与化学计量的量的适当的酸反应来制备。通常在水中或在有机溶剂中,或在这两种溶剂的混合物中进行这样的反应。一般地,当可行时,非水性介质如醚、乙酸乙酯、乙醇、异丙醇或乙腈是优选的。额外合适的盐的列表可见于,例如,Remington's Pharmaceutical Sciences,第20版,Mack Publishing Company,Easton,Pa.,(1985)(其通过引用并入本文)中。
对于临床使用,可单独地或将其配制在药物组合物中,通过有效地实现所需治疗结果的任何合适的施用途径施用本发明的寡核苷酸。施用本发明的寡核苷酸的“途径”是指肠内、肠胃外和局部施用或吸入。本发明的寡核苷酸的肠内施用途径包括口服、经胃、肠道和经直肠。肠胃外途径包括静脉内、腹膜内、肌内、鞘内、皮下、局部注射、经阴道、局部、经鼻、经粘膜和经肺施用。本发明的寡核苷酸的局部施用途径意指将寡核苷酸外部地施加至表皮、颊腔和以及施加至耳、眼和鼻中。
如本文所用,术语“施用”或“施加”旨在包括用于直接或间接地将化合物递送至其期望的作用部位的所有手段。
本文所述的化合物或其药学上可接受的盐和/或水合物可被单独施用,与本发明的其它化合物组合施用,和/或在与其它治疗剂组合的混合物中施用。当然,可与本发明化合物共施用的治疗剂的选择将部分取决于待治疗的病况。
例如,当向具有由依赖于自体诱导物的有机体引起的疾病状态的患者施用时,可在含有用于治疗通常与所述疾病相关的疼痛、感染和其它症状和副作用的试剂的混合物中施用本发明的化合物。这样的试剂包括,例如,镇痛药、抗生素等。
当向经历癌症治疗的患者施用时,可在含有抗癌剂和/或补充增效剂的混合物中施用所述化合物。还可在含有治疗放射疗法的副作用的试剂诸如止吐剂、辐射防护剂等的混合物中施用所述化合物。
可与本发明的化合物共同施用的补充增效剂包括例如,三环抗抑郁药(例如,丙咪嗪、地昔帕明、阿米替林、氯米帕明、曲米帕明、多虑平、去甲替林、普罗替林、阿莫沙平和马普替林);非三环和抗抑郁药(例如,舍曲林、曲唑酮和西酞普兰);Ca+2拮抗剂(例如,维拉帕米、硝苯地平、尼群地平和卡罗维林);两性霉素;曲帕拉醇类似物(例如,他莫昔芬);抗心律失常药物(例如,奎尼丁);降压药(例如,利血平);巯基清除剂(thiol depleter)(例如,丁硫氨酸和亚砜亚胺)以及甲酰四氢叶酸钙。
施用本发明的活性化合物本身,或以其中将活性化合物与一种或多种药学上可接受的载体、赋形剂或稀释剂混合的药物组合物的形式施用本发明的活性化合物。通常使用一种或多种生理上可接受的载体(包括赋形剂和助剂)以常规方式配制用于根据本发明使用的药物组合物,所述载体有利于将活性物质加工成可药用的制剂。适当的制剂取决于所选择的施用途径。
对于经粘膜施用,将适合于待渗透的屏障的渗透剂用于制剂。这样的渗透剂在本领域中是公知的。
对于口服施用,可通过将活性化合物与本领域中公知的药学上可接受的载体组合来容易地配制所述化合物。这样的载体使得本发明的化合物能够被配制成用于待治疗的患者口服摄入的片剂、丸剂、锭剂、胶囊、液体、凝胶、糖浆、浆液、悬浮液。可利用固体赋形剂,任选地通过研磨所得的混合物,和在添加合适的助剂(如果需要的话)后加工颗粒的混合物以获得片剂或锭芯,来获得用于口服使用的药物制剂。合适的赋形剂具体地是填充剂诸如糖,包括乳糖、蔗糖、甘露醇或山梨糖醇;纤维素制剂诸如,例如,玉米淀粉、小麦淀粉、稻淀粉、马铃薯淀粉、明胶、黄蓍胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮(PVP)。如果需要的话,可添加崩解剂,诸如交联的聚乙烯吡咯烷酮、琼脂或藻酸或其盐诸如藻酸钠。
为锭芯提供合适的包衣。为此目的,可使用浓糖溶液,其可任选含有阿拉伯树胶、滑石、聚乙烯吡咯烷酮、卡波姆凝胶、聚乙二醇和/或二氧化钛、漆溶液和合适的有机溶剂或溶剂混合物。可将染料或色素添加入片剂或锭包衣以用于辨识或表征活性化合物剂量的不同组合。
可口服施用的药物制剂包括由明胶制成的推入配合胶囊,以及由明胶和增塑剂诸如甘油或山梨糖醇制成的软密封胶囊。推入配合胶囊可包含与填充剂诸如乳糖、粘合剂诸如淀粉和/或润滑剂诸如滑石或硬脂酸镁和任选地稳定剂混合的活性成分。在软胶囊中,可将活性化合物溶解或悬浮在合适的液体诸如脂肪油、液体石蜡或液体聚乙二醇中。另外,可添加稳定剂。用于口服施用的所有制剂应该以适合用于这样的施用的剂量存在。
对于口腔施用,所述组合物可采取以常规方式配制的片剂或锭剂的形式。
对于通过吸入的施用,通过使用合适的喷射剂例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其它合适的气体,以从加压包装或喷雾器的气溶胶喷雾呈递的方式,方便地递送用于根据本发明使用的化合物。在加压气溶胶的情况下,剂量单位可通过提供阀以递送计量的量来确定。用于在吸入器或吹入器中使用的例如明胶的胶囊和药筒可被配制来含有化合物与合适的粉末基质诸如乳糖或淀粉的粉末混合物。
所述化合物可被配制来通过注射,例如,通过推注或连续输注来进行肠胃外施用。对于本发明的组合物,注射是优选施用方法。可以以单位剂量形式,例如在安瓿或多剂量容器中在具有添加的防腐剂的情况下呈递用于注射的制剂。所述组合物可采取诸如油性或水性媒介物中的混悬剂、溶液或乳液的形式,并可含有可被添加的配制剂诸如悬浮剂、稳定剂和/或分散剂,诸如交联聚乙烯吡咯烷酮、琼脂或海藻酸或其盐诸如藻酸钠。
用于肠胃外施用的药物制剂包括水溶性形式的活性化合物的水溶液。另外,可将活性化合物的悬浮液配制为适当的油性注射悬浮液。合适的亲脂性溶剂或媒介物包括脂肪油诸如芝麻油,或合成脂肪酸酯,诸如油酸乙酯或甘油三酯,或脂质体。水性注射悬浮液可含有增加悬浮液的粘度的物质,诸如羧甲基纤维素钠、山梨醇或葡聚糖。任选地,所述悬浮液还可以含有合适的稳定剂或增加化合物的溶解度以允许高浓度溶液的制备的试剂。对于注射,可在水溶液中,优选在生理上相容的缓冲液诸如Hanks溶液、林格氏溶液或生理盐水缓冲液中配制本发明的试剂。
或者,活性成分可呈粉末形式,用于在使用前利用合适的媒介物例如无菌无热原水进行重建。
还可在直肠组合物诸如栓剂或保留灌肠剂(例如,含有常规栓剂基质诸如可可脂或其它甘油酯)中配制所述化合物。
除了先前描述的制剂以外,还可将所述化合物配制为贮库制剂。这样的长效制剂可通过植入或经皮递送(例如,皮下或肌内)、肌内注射或透皮贴片来施用。因此,例如,可用合适的聚合或疏水材料(例如,在可接受的油中作为乳剂)或离子交换树脂配制所述化合物,或将其配制为微溶性衍生物,例如配制为微溶性盐。
所述药物组合物还可包括合适的固体或凝胶相载体或赋形剂。这样的载体或赋形剂的实例包括碳酸钙、磷酸钙、各种糖、淀粉、纤维素衍生物、明胶和聚合物诸如聚乙二醇。
优选的药物组合物是被配制来用于注射诸如静脉内注射的组合物,并且包含按重量计约0.01%至约100%的本发明的化合物(基于100%的总的药物组合物的重量)。药物-配体缀合物可以是抗体-细胞毒素缀合物,其中抗体已被选择来靶向特定癌症。
在一些实施方案中,本发明的药物组合物还包含附加治疗剂。
在一些实施方案中,所述附加治疗剂是抗癌剂。
在一些实施方案中,所述附加抗癌剂选自抗代谢物、拓扑异构酶I和II的抑制剂、烷化剂、微管抑制剂、抗雄激素剂、GNRh调节剂及其混合物。
在一些实施方案中,附加治疗剂是化疗剂。
本文中的“化疗剂”意指可用于癌症治疗的化合物。实例为(但不限于):吉西他滨、伊立替康、多柔比星、5-氟尿嘧啶、胞嘧啶阿拉伯糖苷("Ara-C")、环磷酰胺、塞替派、白消安、细胞毒素、TAXOL、甲氨蝶呤、顺铂、美法仑、长春碱和卡铂。
在一些实施方案中,第二化疗剂选自他莫昔芬、雷洛昔芬、阿那曲唑、依西美坦、来曲唑、伊马替尼、紫杉醇、环磷酰胺、洛伐他汀、含羞草素(minosine)、吉西他滨、阿糖胞苷、5-氟尿嘧啶、甲氨蝶呤、多西他赛、戈舍瑞林、长春新碱、长春碱、诺考达唑、替尼泊苷、依托泊苷、吉西他滨、埃坡霉素、长春瑞滨、喜树碱、柔红霉素、放线菌素D、米托蒽醌、吖啶、多柔比星、表阿霉素或伊达比星。
试剂盒
在另一个方面,本发明提供了含有一种或多种本发明的化合物或组合物和使用所述化合物或组合物的说明书的试剂盒。在示例性实施方案中,本发明提供了用于将本发明的接头臂缀合于另一个分子的试剂盒。所述试剂盒包括接头,和用于将所述接头附接于特定官能团的说明书。所述试剂盒还可包括一种或多种细胞毒性药物、靶向剂、可检测的标记物、药物盐或缓冲剂。所述试剂盒还可包括容器和任选地一个或多个小瓶、试管、烧瓶、瓶或注射器。试剂盒的其它形式对于本领域技术人员将是显而易见的,并且在本发明的范围之内。
医学用途
因此,在另一个方面,本发明提供了抑制MYD88 L265P阳性肿瘤/癌症细胞的增殖的方法,其包括向肿瘤细胞施用本发明的化合物。在一些实施方案中,肿瘤可以转移性的或非转移性的。
本文中的“癌症”或“肿瘤”意指其特征在于失调的细胞增殖的人中的病理状态。实例包括但不限于:癌、淋巴瘤、母细胞瘤和白血病。癌症的更具体的实例包括但不限于:肺癌(小细胞和非小细胞癌)、乳腺癌、前列腺癌、类癌、膀胱癌、胃癌、胰腺癌、肝癌(肝细胞癌)、肝母细胞瘤、结直肠癌、头颈鳞状细胞癌、食道癌、卵巢癌、子宫颈癌、子宫内膜癌、间皮瘤、黑素瘤、肉瘤、骨肉瘤、脂肪肉瘤、甲状腺癌、硬纤维瘤、急性髓细胞性白血病(AML)和慢性髓细胞性白血病(CML)。
本文中的“抑制”或“治疗”或“医治”意指减轻、治疗性治疗和预防性或防治性治疗,其中目的是减轻或防止目标病理病症或病况。在一个实例中,在施用本发明的化合物后,癌症患者可经历肿瘤尺寸的减小。“治疗”或“医治”包括(1)抑制正在经历或表现出疾病的病理学或症状的受试者的疾病,(2)改善正在经历或表现出疾病的病理学或症状的受试者的疾病,和/或(3)实现正在经历或表现出疾病的病理学或症状的受试者或患者的疾病的任何可测量的减轻影响的受试者或患者正在经历或显示病理学或疾病的症状中的疾病的任何可测量的减轻。在本发明的化合物可阻止癌细胞生长和/或杀死癌细胞的这个意义上,其可以是细胞生长抑制性的和/或具有细胞毒性的。
本文中的“治疗有效量”意指对于“治疗”受试者或哺乳动物的病症是有效的本文中提供的化合物的量。在癌症的情况下,治疗有效量的药物可减少癌细胞的数量,减小肿瘤尺寸,抑制癌细胞浸润至周围器官中,抑制肿瘤转移,抑制肿瘤生长至一定程度,和/或缓解与癌症相关的一个或多个症状至一定程度。
在另一个方面,本发明提供了治疗受试者的MYD88 L265P阳性肿瘤/癌症的方法,其包括向受试者施用治疗有效量的本发明的化合物。在一些实施方案中,所述肿瘤或癌症可处于任何阶段,例如,早期或晚期,例如I期、II期、III期、IV期或V期的肿瘤或癌症。在一些实施方案中,所述肿瘤或癌症可以是转移性或非转移性的。在转移的情况下,本发明的方法可减轻或抑制原发肿瘤或癌症至其它部位的转移,或转移性肿瘤或癌症在远离原发肿瘤或癌症治疗的其它部位的形成或建立。因此,本发明的方法包括,除其以外,1)减少或抑制潜在地或确实发生转移的肿瘤或癌细胞(例如,散播的肿瘤细胞,DTC)的生长、增殖、迁移性或侵袭性;2)减少或抑制从原发性肿瘤或癌症产生的至一个或多个与原发性肿瘤或癌症不同的其它部位、位置或区域的转移瘤的形成或建立;3)在转移瘤已形成或已建立后减少或抑制在与原发性肿瘤或癌症不同的一个或多个其它部位、位置或区域的转移瘤的生长或增殖;和4)在转移瘤已形成或建立后减少或抑制另外的转移瘤的形成或建立。
在一些实施方案中,所述肿瘤或癌症是实体或液体细胞团。“实体”肿瘤是指通常聚集在一起并形成团块的癌症、肿瘤或转移瘤。具体的非限制性实例包括乳腺、卵巢、子宫、子宫颈、胃、肺、胃部、结肠、膀胱、神经胶质和子宫内膜肿瘤/癌症等。“液体肿瘤”,其是指天然分散或弥漫的肿瘤,因为它们通常不形成实体团块。具体的实例包括网状内皮或造血系统的肿瘤,诸如淋巴瘤、骨髓瘤和白血病。白血病的非限制性实例包括急性和慢性成淋巴细胞瘤、成髓细胞瘤和多发性骨髓瘤。通常地,这样的疾病产生自不良分化的急性白血病,例如,成红细胞白血病和急性巨核细胞白血病。具体的骨髓病症包括,但不限于,急性早幼粒细胞白血病(APML)、急性骨髓性白血病(AML)和慢性骨髓性白血病(CML)。淋巴样恶性肿瘤包括,但不限于,急性淋巴细胞白血病(ALL),其包括B-谱系ALL(B-ALL)和T谱系ALL(T-ALL)、慢性淋巴细胞白血病(CLL)、幼淋巴细胞白血病(PLL)、毛细胞白血病(HLL)和Waldenstroem巨球蛋白血症(WM)。具体的恶性淋巴瘤包括非何杰金氏淋巴瘤及其变型、外周T细胞淋巴瘤、成人T细胞白血病/淋巴瘤(ATL)、皮肤T细胞淋巴瘤(CTCL)、大颗粒淋巴细胞白血病(LGF)、何杰金氏病和Reed-Sternberg病。
在一些实施方案中,可将本发明的方法与其它治疗或疗法(例如,手术切除、放射疗法、电离或化学放射疗法、化学疗法、免疫疗法、局部或区域热(高热)疗法或疫苗接种)一起实施。可在施用本发明的化合物之前,基本上与之同时(单独地,或在混合物中)或之后施用这样的其它治疗或疗法。
在一些实施方案中,本发明的方法包括施用治疗有效量的与附加治疗剂组合的本发明的化合物。在一些实施方案中,所述附加治疗剂是抗癌剂/抗肿瘤剂。在一些实施方案中,所述附加治疗剂是抗代谢物、拓扑异构酶I和II的抑制剂、烷化剂、微管抑制剂、抗雄激素剂、GNRh调节剂或其混合物。在一些实施方案中,附加治疗剂选自他莫昔芬、雷洛昔芬、阿那曲唑、依西美坦、来曲唑、伊马替尼、紫杉醇、环磷酰胺、洛伐他汀、含羞草素、吉西他滨、阿糖胞苷、5-氟尿嘧啶、甲氨蝶呤、多西他赛、戈舍瑞林、长春新碱、长春碱、诺考达唑、替尼泊苷、依托泊苷、吉西他滨、埃坡霉素、长春瑞滨、喜树碱、柔红霉素、放线菌素D、米托蒽醌、吖啶、多柔比星、表阿霉素或伊达比星。
与一种或多种附加治疗剂“组合”的施用包括以任意顺序同时(共同)和相继施用。如本文所用,术语“药物组合”是指从混合或组合活性成分获得的产品,其包括活性成分的固定和非固定组合。术语“固定组合”意指活性成分,例如式(1)的化合物和共剂,均以单一实体或剂量的形式同时施用于患者。术语“非固定组合”指活性成分,例如式(1)的化合物和共剂,均作为单独实体同时、共同或相继地(无特定时间限制)施用至患者,其中这样的施用在患者的体内提供了治疗有效水平的活性成分。后者还用于鸡尾酒疗法,例如三种或更多种活性成分的施用。
有效剂量
适合与本发明一起使用的药物组合物包括其中以治疗有效量(即,以对于实现其期望的目的是有效的量)含有活性成分的组合物。对于特定应用有效的实际量将取决于,除其他以外,待治疗的病况。有效量的确定完全在本领域技术人员(尤其地根据本文中的详述的公开内容)的能力之内。
寡核苷酸之一的“治疗有效量”是指用于获得治疗或预防受试者的免疫介导的病症诸如B细胞淋巴瘤的期望结果的寡核苷酸的充足量。可将本发明的寡核苷酸以纯的形式使用或在药学上可接受的载体中使用。或者,可将寡核苷酸作为药物组合物来施用。本发明中的“量”应指剂量。所述剂量可通过本领域技术人员公知的标准技术来确定,并且可取决于因素(包括,但不限于受试者的身型和/或总体健康或疾病的严重程度)而变化。可作为单次治疗或在一系列治疗中进行本发明的寡核苷酸的导入。用于施用的本发明的寡核苷酸的受试剂量在每次施用约1μg至100mg的范围内。然而,可在为上述剂量的10倍至1000倍的范围内使用用于免疫介导的病症的治疗的剂量。可由本领域技术人员,例如由主治医生在合理的医学判断范围内调整更优选的剂量以提供最佳治疗效果。可在一个或多个预防性或治疗性施用中施用治疗有效量。当应用于单独施用的个体活性成分时,该术语是指该单独的成分。当应用于组合时,该术语是指导致治疗效果的活性成分的组合量,无论是相继还是同时进行的组合施用。
对于本文所述的任何化合物,治疗有效量最初可以从细胞培养测定来确定。目标血浆浓度将是能够抑制细胞生长或分裂的活性化合物的那些浓度。在优选实施方案中,细胞活性被抑制至少25%。能够诱导至少约30%、50%、75%或甚至90%或更高的细胞活性抑制的活性化合物的目标血浆浓度目前是优选的。可监测患者中细胞活性的抑制百分比以评估所获得的血浆药物浓度的适宜性,并且可向上或向下调整该剂量,以达到所需的抑制百分比。
正如本领域中所公知的,用于人的治疗有效量也可从动物模型来确定。例如,用于人的剂量可被配制来达到已被发现在动物中是有效的循环浓度。人中的剂量可以通过监测细胞抑制以及向上或向下调整剂量来进行调整,如上所述的。
治疗有效剂量也可从关于已知表现出类似的药理学活性的化合物的人数据来确定。可基于所施用的化合物相较于已知的化合物的相对生物利用度和效力来调整施用剂量。
基于上述方法和如本领域中公知的其它方法调整剂量以在人中实现最大功效完全在本领域普通技术人员的能力之内。
在局部施用的情况下,所施用的化合物的全身性循环浓度不会是特别重要的。在这种情况下,施用所述化合物以在局部区域获得有效实现所期望的结果的浓度。
为了用于预防和/或治疗与异常细胞增殖相关的疾病,约0.001μM至20μM的施用的化合物的循环浓度是优选的,其中约0.01μM至5μM是优选的。
用于本文所述的化合物的口服施用的患者剂量通常在约1mg/天至约10000mg/天,更常见地约10mg/天至约1000mg/天,最常见地约50mg/天至约500mg/天的范围内。按照患者体重来说,典型的剂量范围为约0.01至约150mg/kg/天,更常见地为约0.1至约15mg/kg/天,最常见地为约1至约10mg/kg/天,例如5mg/kg/天或3mg/kg/天。
在至少一些实施方案中,延迟或抑制肿瘤生长的患者剂量可以是1μmol/kg/天或更小。例如,患者剂量可以是0.9、0.6、0.5、0.45、0.3、0.2、0.15或0.1μmol/kg/天或更少(参照药物的摩尔数)。优选地,抗体与药物的缀合物,当在至少5天的一段时间中以日剂量的量施用时,延迟肿瘤的生长。
对于其它施用方式,可单独地调整剂量和间隔时间,以提供对于待治疗的特定临床适应症是有效的所施用的化合物的血浆水平。例如,在一个实施方案中,可每天以相对高的浓度多次施用根据本发明的化合物。或者,可能更加期望以最小的有效浓度施用本发明的化合物以及使用不太频繁的施用方案。这将提供与个体的疾病的严重度相称的治疗方案。
通过利用本文提供的教导,可规划有效的治疗性治疗方案,所述方案不引起实质性的毒性,但对于治疗由特定患者表现出的临床症状是完全有效的。该规划应牵涉通过考虑因素诸如化合物的效力、相对生物利用度、患者体重、不良副作用的存在和严重度、优选施用模式和所选择的试剂的毒性特征谱来进行活性化合物的仔细选择。
虽然在本文中已显示和描述了本发明的优选实施方案,但对于本领域技术人员来说很显然的是,这样的实施方案仅通过举例的方式提供。对于本领域技术人员来说,现可进行许多变化、改变和替换而不脱离本发明的范围。应当理解的是,可在实施本发明中应用对本文所述的本发明的实施方案的各种替代。下文的权利要求旨在限定本发明的范围并且这些权利要求范围内的方法和结构以及它们的等同物被其涵盖。
可单独地,与它们自身组合地,在药学上可接受的载体中,与一种或多种附加活性成分组合地使用本发明的寡核苷酸。本发明的寡核苷酸和附加活性成分的施用可以是相继的或同时的。所述活性成分包括非类固醇抗炎剂、类固醇、非特异性免疫抑制剂、生物反应调节剂、化学化合物、小分子、核酸分子和TLR拮抗剂。活性成分还指通过拮抗趋化因子,通过诱导调节性T细胞(CD4+CD25+T细胞)的产生,通过抑制补体、基质金属蛋白酶和一氧化氮合酶,通过阻断共刺激因子和通过抑制免疫细胞中的信号传导级联抑制免疫激活的试剂。非类固醇抗炎剂包括但不限于,双氯芬酸、二氟尼柳、依托度酸、氟比洛芬、布洛芬、吲哚美辛、酮洛芬、酮咯酸、萘丁美酮、萘普生、奥沙普秦、吡罗昔康、舒林酸、tohnetin、塞来考昔和罗非考昔。类固醇包括,但不限于,可的松、地塞米松、氢化可的松、甲泼尼龙、泼尼松龙、泼尼松和曲安西龙。非特异性免疫抑制剂意指用于阻止免疫介导的病症的发展的试剂。非特异性免疫抑制剂包括但不限于环磷酰胺、环孢菌素、甲氨蝶呤、类固醇、FK506、他克莫司、霉酚酸和西罗莫司。生物反应调节剂包括重组白介素-1受体拮抗剂(Kineret或anakima)、可溶性p75TNFα受体-IgG1融合蛋白(依那西普或Enbrel),或针对TNFα的单克隆抗体(英夫利昔单抗或RemicadeX)。试剂还包括干扰素β-1a、白细胞介素-10和TGFβ。
可在递送媒介物中或与递送媒介物一起,或以与媒介物连接的形式施用本发明的寡核苷酸。所述媒介物包括,但不限于,固醇(例如,胆固醇)、脂质卷(cochleates)、乳脂体、ISCOM;脂质(例如,阳离子脂质、阴离子脂质)、脂质体;乙二醇(PEG);活细菌载体(例如,沙门菌属(Salmonella)、大肠杆菌、卡介苗、志贺菌属(Shigella)、乳杆菌属(Lactobacillus))、活病毒载体(例如,牛痘病毒、腺病毒、单纯疱疹)、病毒体、病毒样颗粒、微球、核酸疫苗、聚合物(例如,羧甲基纤维素、壳聚糖)、聚合物环和通过特异性受体识别靶细胞的靶向剂。
聚乙二醇化是将聚(乙二醇)聚合物链共价附接于另一种分子(通常为药物或治疗性蛋白)的过程。通过将PEG的反应性衍生物与靶试剂一起温育来常规地实现聚乙二醇化。聚乙二醇化试剂可“掩蔽”试剂而免受宿主的免疫系统攻击,增加试剂的水动力学尺寸(这延长其循环时间)。可聚乙二醇化本发明的寡核苷酸。
药学上可接受的载体是指一种或多种适合用于向受试者施用本发明的寡核苷酸的固体或液体填充剂、稀释剂或包封物质。所述载体可以是有机的、无机的、天然或合成的。所述载体包括任何和所有溶液、稀释剂、溶剂、分散介质、脂质体、乳剂、包衣、抗菌和抗真菌剂,等渗和吸收延迟剂,以及适于用于施用本发明的寡核苷酸的任何其它载体,并且它们的用途在本领域是公知的。根据寡核苷酸的具体施用模式来选择药学上可接受的载体。肠胃外制剂通常包含可注射流体,其包括药学上和生理上可接受的流体诸如水、生理盐水、平衡盐溶液、葡萄糖水、甘油等作为媒介物。对于固体组合物(例如,粉剂、丸剂、片剂或胶囊形式),常规的无毒固体载体可包括,例如,药物级的甘露糖醇、乳糖、淀粉或硬脂酸镁。除了生物中性载体以外,待施用的药物组合物还可含有少量的无毒辅助物质,如润湿剂或乳化剂、防腐剂和pH缓冲剂等,例如乙酸钠或脱水山梨醇单月桂酸酯。
在一些实施方案中,通过口服、肠内、肠胃外或局部使用的途径或吸入施用所述寡核苷酸。
联合治疗
在另一个方面,本发明提供用于联合治疗的组合物和方法,所述联合治疗将寡核苷酸与化疗剂诸如Btk抑制剂、PI3Kδ抑制剂、IRAK抑制剂、抗-CD20单克隆抗体、SYK抑制剂或Bcl-2抑制剂组合施用。
“化疗剂”是可用于癌症治疗的化合物。实例是,但不限于:吉西他滨、伊立替康、多柔比星、5-氟尿嘧啶、胞嘧啶阿拉伯糖苷("Ara-C")、环磷酰胺、塞替派、白消安、细胞毒素、TAXOL、甲氨蝶呤、顺铂、美法仑、长春碱和卡铂。
与一种或多种其它治疗剂“结合”施用包括同时(共同)和以任何顺序相继施用。如本文中所用,术语“药物组合”是指从混合或组合活性成分获得的产品,并且包括活性成分的固定和非固定的组合。术语“固定组合”意指活性成分,例如本发明的ODN和共剂,均以单一实体或剂量的形式同时施用至患者。术语“非固定的组合”意指活性成分,例如本发明的ODN和共剂,均作为单独的实体被同时地、共同地或在无特定时间限制的情况下相继地被施用至患者,其中此种施用在患者体内提供了治疗有效水平的活性成分。后者还适用于鸡尾酒疗法,例如三种或更多种活性成分的施用。此种联合治疗还可包括不止一次的根据本发明的化合物和/或独立地其它试剂的单次施用。根据本发明的化合物和其它试剂的施用可以通过相同或不同的途径。
B细胞受体(BCR)是位于B细胞的外表面上的跨膜受体蛋白。当B细胞因其与结合其受体的抗原的第一次相遇而被活化时,所述细胞增殖和分化,以产生分泌抗体的浆B细胞和记忆B细胞的群体。BCR在与Ag相互作用后具有两个关键功能。一个功能是信号传导,涉及受体寡聚的变化。第二个功能是介导内化用于Ag的随后处理和肽至辅助T细胞的呈递。BCR功能是正常的抗体产生所需的,并且BCR信号传导中的缺陷可导致免疫缺陷、自身免疫和B细胞恶性肿瘤(Corcos D.等,Blood 117(26):6991–8)。B细胞受体可执行几个信号传导途径,包括BTK、PI3K和IKK/NF-κB信号传导途径(Tomohiro K等Annual Review of Immunology 2008 28(1):21)。B细胞受体信号传导目前是各种淋巴样肿瘤的治疗靶。
最近,BTK已成为用于治疗B谱系白血病和淋巴瘤的新的抗凋亡分子靶。BTK主要在B淋巴细胞、淋巴细胞前体和发育中的骨髓细胞上表达(Smith CI等J Immunol.1994 557–565),并且在慢性BCR活化中是必需的(Davids MS等Leuk Lymphoma.2012 2362–2370;Dal Porto JM等Mol Immunol.2004 599–613)。在BCR活化后,PI3K被激活,这进而刺激磷酸肌醇-3,4,5-(PIP3)的产生。一旦产生足够量的PIP3,BTK就被募集至质膜,随后通过Src家族激酶,尤其是LYN和FYN在Y551位点上经历磷酸化(Afar DE等Mol Cell Biol.1996 3465–3471;Winer ES等Expert Opin Investig Drugs.2012 355–361)。磷酸化的BTK激活磷脂酶Cy2,导致蛋白激酶(诸如蛋白激酶C-β)的下游激活和,最后,转录因子NFκB的激活。因此,IRAK及BTK似乎独立地指导下游NF-κB激活,并且IRAK与BTK抑制剂的组合使用已被证明在MYD88中导致协同的肿瘤细胞杀伤(Yang G等2012JCO 8106)。Ibrutinib(PCI-32765)(一种首创BTK抑制剂)已在临床开发中显示有前途的功效。约40%的已复发的或对于治疗是难治性的ABC DLBCL患者对ibrutinib起反应。但其似乎在具有GCB亚型的患者中无作用(Advani RH等J Clin Oncol.2013 31:88-94),这意味着MYD88突变可能在ABC DLBCL患者响应治疗中起着重要作用。另外,有许多研究是探索BTK抑制剂与其它治疗组合的协同功效。例如,显示与利妥昔单抗组合的ibrutinib的II期研究具有显著活性(ORR=85%),并缩短了具有高危特性的CLL患者中重新分布淋巴细胞增多的持续时间(Burger J,Proc ASH Abst.187)。
如上所述,PI3K在TLR和BCR信号传导途径中起着重要作用。PI3K是这样的脂质激酶,其催化质膜的内叶内的磷酸肌醇-3,4,5-三磷酸的产生,从而产生针对许多细胞内含普列克底物蛋白同源结构域的效应子的结合位点,其触发控制细胞分裂、存活、新陈代谢、细胞内运输、分化、肌动蛋白细胞骨架的重新组织和细胞迁移的信号传导级联(Vanhaesebroeck B等Rev Mol Cell Biol.2010 329-341;Hawkins PT等Biochem Soc Trans.2006 647-662)。因此,PI3K信号的抑制可以减少细胞增殖,并且在一些情况下,促进细胞死亡。1A类PI3K酶是包含调节亚基(p85)和催化亚基(p110)的异二聚体(Carpenter CL等J Biol.Chem.1900 19704–11)。p110α和p110β被广泛表达,而p110γ和p110δ主要在白细胞中表达以及在非白细胞来源的一些癌细胞系和人组织诸如乳腺癌细胞中以高水平表达(Hu P等Mol Cell Biol.1993 7677-88;Chantry D等Biol Chem.1997 19236-41;Sawyer C等Cancer Res.2003 1667-75)。据报道,p110δ有助于B细胞的发育及维持、转化和增殖(Hammer SB等J.Leukocyte Bio.2010 1082-1095)。p110δPI3K因BCR信号传导和由来自肿瘤微环境的因子提供的其它信号的改变而在B细胞恶性肿瘤中被过度活化(Pauls SD等Front Immunol.2012 224-229)。已在来自患有慢性淋巴细胞白血病(CLL)、多发性骨髓瘤(MM)细胞系和何杰金氏淋巴瘤(HL)的患者的细胞中已测定了较高水平的p110δPI3K活性(Herman SE等Blood.2010 2078-88;Ikeda H等Blood.2010 1460-68;Meadows SA等Blood.2012 1897-1900)。
p110δ在B细胞中的关键作用导致高度p110δ特异性抑制剂的开发,包括最初开发的用于治疗B细胞恶性肿瘤的idelalisib(CAL-101/GS-1101)(Lannutti BJ等Blood 2011 591-4)。已在来自不同B细胞恶性肿瘤包括CLL和DLBCL的细胞系和患者细胞中研究了idelalisib和其它p110δ选择性抑制剂的活性(Ikeda H等Blood.2010 1460-8;Hoellenriegel J等Blood.2011 591-494)。已证明idelalisib对p110δ的抑制作用降低了由B细胞分子驱动的B-CLL存活,并且通过阻断细胞进入保护性小生境,从而抑制否则会促进B细胞存活和增殖的环境相互作用来进一步起作用。事实上,已发现B-CLL中的Akt的磷酸化以及CXCL13、CCL3、CCL4和TNFα的血浆水平在利用idelalisib治疗的患者中显著降低,这表明p110δ的抑制破坏了B-CLL与它们的保护性微环境的相互作用。最近,与利妥昔单抗组合的idelalisib(作为CLL的治疗)已在无进展存活(PFS)的主要终点上显示高度地统计上显著的延长(2013年ASH第55届年会)。
利妥昔单抗是首次被美国FDA批准为被设计来治疗B细胞恶性肿瘤的抗肿瘤剂的抗CD20单克隆抗体。CD20是在前B淋巴细胞和成熟B淋巴细胞上被特异性发现的细胞表面标志物,并且未被发现存在于其它细胞类型上和游离于循环中(Maloney DG等Blood 1994 2457–2466)。利妥昔单抗与位于B淋巴细胞上的细胞表面CD20的结合导致淋巴细胞的破坏,以治疗许多淋巴瘤、白血病、移植排斥和自身免疫疾病。在临床上,单独的利妥昔单抗对非何杰金氏淋巴瘤(NHL)例如DLBCL的治疗是有效的(McLaughlin P等J Clin Oncol.1998 2825–33;Sano T等International J Clin Oncol.2007 59-62)。重要地,与环磷酰胺、多柔比星、长春新碱和泼尼松(CHOP)治疗组合的利妥昔单抗在DLBCL和许多其它B细胞淋巴瘤的治疗中优于单独的CHOP(Hiddemann W等Blood 2005 3725–3732)。此外,向NHL的标准利妥昔单抗-CHOP化疗添加ibrutinib导致100%的目标响应率(Younes A等J Clin Oncol.2013年第31卷,增刊abs.8502)。其它抗CD20治疗,包括维妥珠单抗(靶向CD20受体的人源化单克隆抗体的丸剂形式),在临床试验中用于治疗非何杰金氏淋巴瘤(NHL)和自身免疫性疾病(Milani C等Curr Opin Mol Ther.2009 200-207)。
已知SYK在传递维持信号和BCR的活化中起着重要作用。在通过抗原的BCR连接后,Lyn PTK被激活并且磷酸化受体的胞质尾中的免疫受体酪氨酸基活化(ITAM)基序。随后磷酸化的ITAM与Syk的SH2结构域的缔合导致Syk通过自身磷酸化的激活。Syk自磷酸化是免疫受体介导的Syk的激活所必需的,这是信号传导分子(包括磷酸肌醇3-激酶、丝裂原活化蛋白激酶、Ras信号途径、磷脂酶C-γ2活化和钙动员)的级联的活化所必需的(Tamir I等1998Oncogene 1353;Beitz LO等JBC 1999 32662–32666)。Syk激酶的抑制代表用于过敏性和抗体介导的自身免疫性疾病的治疗以及用于淋巴瘤的治疗的有希望的方法(Ulanova M等Expert Opin Ther Targets 2005 901-921;Friedberg JW等Blood.2010 2578–2585)。临床试验中最引人注意的Syk抑制剂由R112、R406、R788和R343代表。它们是在Rigel Pharmaceuticals,Inc开发的强效和选择性ATP竞争性Syk激酶抑制剂。其中,R406能够抑制滋补BCR信号传导,并在某些但非全部原发性DLBCL中诱导凋亡(Chen L等Blood 2008 2230-2237)。口服活性抑制剂R788(福他替尼)在类风湿性关节炎的治疗中引起疾病严重度的剂量依赖性改善,并且在NHL和CLL中具有显著的临床活性(Friedberg JW等Blood 2578–2585)。反义寡核苷酸(ASO)或RNA干扰(RNAi)对Syk激酶表达的抑制是用于治疗炎症性疾病的Syk抑制剂的设计的另一种方法(Saitoh S等Immunity 2000 525-35)。另外,较新的Syk抑制剂诸如GS-9973也处于用于治疗NHL和CLL的临床开发中(Sharman JP等2013ASH Poster 1634)。
通过异常激活的BCR进行的信号传导在某些类型的B细胞肿瘤的发病机理中起着关键作用。如上所述,BTK和SYN均参与BCR信号传导途径。阻断BCR促存活途径在NHL和CLL中均具有极大的治疗潜力。事实上,在过去的几年中,兴奋一直在建立BCR信号传导在CLL和其它B细胞淋巴增生性病症中的临床和治疗重要性。目前,用于治疗CLL的最活跃的治疗方案是常规化疗与单克隆抗CD20抗体利妥昔单抗一起的组合,其具有优良的总体响应率(Seiffert M等Blood 2011 3016-3024)。然而,疗效在因染色体17p的缺失或p53的体细胞突变而丧失正常p53活性的患者的亚组中被削弱(Zenz T等J Clin Oncol.2010 4473-4479)。另外,这些方案与因骨髓抑制和免疫抑制而导致的显著发病率相关。此种毒性往往导致剂量减少或治疗的中断并且不鼓励在老年患者中使用。因此,需要可为患有高风险疾病的患者带来益处以及具有有利的治疗指数的新的治疗。
联合治疗的一个策略是协同地同时靶向TLR和BCR途径。该策略与ABC DLBCL细胞系的存活需要通过TLR和BCR两者的信号的观察一致,因为敲低BCR信号传导室的CD79分子协同地杀死具有MYD88敲低的ABL-DLBCL细胞系(Ngo VN等Nature.2011 115–119)。因此,预期使用TLR9拮抗剂和BCR途径抑制剂(诸如ibrutinib(BTK抑制剂)、利妥昔单抗(抗CD20抗体)、idelalisib(PI3K)和R406(SYK抑制剂))的组合协同杀死具有L265P MYD88的淋巴样肿瘤在CLL患者中改善总体响应率和低毒性。
虽然在本文中已显示和描述了本发明的优选实施方案,但对于本领域技术人员来说显而易见的是,这样的实施方案仅以举例说明的方式提供。许多变化、改变和替换对于本领域技术人员来说现将发生而不脱离本发明的范围。应当理解的是,对本文中描述的本发明的实施方案的各种替代方案可用于实施本发明。下文列权利要求旨在限定本发明的范围并且这些权利要求范围内的方法和结构以及其等同物由此被涵盖。
实施例
通过但不限于以下举例说明本发明的化合物的制备的实施例来进一步举例说明本发明。
在Takara Co.(Dalian,China)合成用于本实施例的以下寡核苷酸(ODN)。TLR9刺激性ODN为:
CpG2395(5’-tcgtcgttttcggcgcgcgccg-3’,SEQ ID NO.:17),
CpG1826(5’-tccatgacgttcctgacgtt-3’,SEQ ID NO.:18),
CpG2216(5’-gggggacgatcgtcgggggg-3’,SEQ ID NO.:19)。
用于本实施例的其它ODN为:
(CCT)6(5’-cctcctcctcctcctcct-3’,SEQ ID NO.:15),
(CCT)7(5’-cctcctcctcctcctcctcct-3’,SEQ ID NO.:16),
(CCT)8(5’-cctcctcctcctcctcctcctcct-3’,SEQ ID NO.:20),
(CCT)8C(5’-cctcctcctcctcctcctcctcctc-3’,SEQ ID NO.:1),
(CCT)8CC(5’-cctcctcctcctcctcctcctcctcc-3’,SEQ ID NO.:2),
(CCT)9(5’-cctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:3),
(CCT)10(5’-cctcctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:6),
(CCT)10C(5’-cctcctcctcctcctcctcctcctcctcctc-3’,SEQ ID NO.:7),
(CCT)10CC(5’-cctcctcctcctcctcctcctcctcctcctcc-3’,SEQ ID NO.:8),
(CCT)11(5’-cctcctcctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:9),
(CCT)11C(5’-cctcctcctcctcctcctcctcctcctcctcctc-3’,SEQ ID NO.:10),
(CCT)11CC(5’-cctcctcctcctcctcctcctcctcctcctcctcc-3’,SEQ ID NO.:11),
(CCT)12(5’-cctcctcctcctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:12),
(CCT)14(5’-cctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:13)和
(CCT)16(5’-cctcctcctcctcctcctcctcctcctcctcctcctcctcctcctcct-3’,SEQ ID NO.:14)。
将在以下实施例中用于处理寡核苷酸(ODN)的所有试剂稀释在PBS中,并通过Limulus阿米巴样细胞裂解物测定(Accumedi Solutions Co.,Ltd,Zhanjiang,China)测试内毒素。
实施例1
通过测序在MYD88 L265P突变体存在的情况下确认弥漫性大B细胞淋巴瘤(DLBCL)细胞系
实验方法
将具有MYD88 L265P突变体的OCI-Ly3.3和MYD88野生型OCI-Ly19培养在补充有100mg/mL青霉素/链霉素以及10%和20%胎牛血清的Iscove改良Dulbecco培养基(IMDM,Hyclone,Logan,UT,USA)中。将所有细胞于潮湿的条件下保持在37℃5%CO2中。使用基因组DNA试剂盒(TransGen Biotech Co.Beijing,China)从3×106个OCI-Ly3.3或OCI-Ly19细胞提取细胞DNA,并使用Tks Gflex DNA聚合酶(Takara Biotechnology,Dalian,China)以及MYD 88正向引物(5’-GTTGAAGACTGGGCTTGTCC-3’,SEQ ID NO.:51)和反向引物(5’-AGGAGGCAGGGCAGAAGTA-3’,SEQ ID NO.:52)进行聚合酶链式反应(PCR)。通过凝胶提取试剂盒(Kangwei Biotechnology,Beijing,China)提取PCR产物,并将其克隆入pEasy-Blunt克隆试剂盒(TransGen Biotech Co.Beijing,China)。来自每一个细胞系的两个克隆被选择用于由Genwiz Co.(Beijing,China)测序以检测MYD 88 L265P突变。
实验结果
确认了OCI-Ly3.3的MYD88 L265P突变体的存在(图1,右),并且OCI-Ly19不具有该突变体(图1,左)。与先前的报导一致,OCI-Ly3.3具有纯合的MYD88 L265P突变体(图1,右)。
实施例2
TLR7/TLR9拮抗剂对具有MYD88 L265P突变体的ABC-DLBCL细胞的作用
实验方法
为了观察TLR7/9拮抗剂对ABC-DLBCL细胞增殖的抑制作用,以下文中指定的剂量范围,用TLR7/TLR9拮抗剂培养96孔板中的5x 105/孔的OCI-Ly3.3和OCI-Ly19细胞。利用购自Beyotime Institute of Biotechnology(Jiangsu,China)的基于四唑盐的测定(WST-1)测量细胞活力。将活细胞的百分比计算为450nm处的处理的细胞的吸光度对对照细胞的吸光度的比率。
为了利用TLR7/TLR9拮抗剂探究细胞因子从DLBCL细胞的分泌,将96孔板中的2x 105/孔的OCI-Ly19或OCI-Ly 3.3细胞与具有下文中标示的浓度的TLR7/9拮抗剂(CCT)8、(CCT)12和(CCT)12-M(分别对应于NO.20、12和43的序列ID)一起温育。在36小时的温育后通过细胞计数珠测定(cytometry beads assay)分析上清液中的细胞因子IL-10。
实验结果
TLR7/9拮抗剂能够抑制具有MYD88 L265P突变的OCI-Ly3.3细胞的存活,但不能抑制野生型细胞的存活。
媒介物的最大终浓度不对测试的细胞系诱导任何细胞毒性(数据未显示)。0.3uM和1uM的(CCT)8、(CCT)12和(CCT)12-M导致对OCI-Ly3.3的抑制(图2B),但不导致对OCI-Ly19细胞的抑制(图2A)。门控是CD19(或CD3)的FACS点图和侧向散射(SSC)。将门设置在CD19(或CD3)阳性细胞上,并且结果代表CD19或CD3阳性细胞的百分比。
降低的OCI-Ly3.3细胞活力与细胞因子的抑制相关。
图3显示所有3种TLR7/9拮抗剂能够抑制OCI-Ly3.3细胞中的IL-10分泌。此处显示的数据是来自3个独立实验的平均值。在MYD88L265野生型OCI-Ly19细胞中不能检测到IL-10(数据未显示)。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于短链核酸探针和双链特异性核酸内切酶的高特异性microRNA荧光检测方法与流程
- IL‑1‑α和‑β双特异性双重可变结构域免疫球蛋白及其用途的制造方法与工艺
- 一种特异性标记家蚕微孢子虫的核酸探针及其原位荧光杂交检测方法与流程
- 核酸的等位基因特异性扩增的制造方法与工艺
- 一种特异性检测弓形虫核酸的荧光PCR方法和试剂盒与流程
- 一种特异性检测人类微小病毒B19核酸的荧光PCR方法和试剂盒与流程
- 一种特异性结合新城疫病毒的核酸适配体及其筛选方法和应用与流程
- 球形核酸纳米颗粒缀合物的序列特异性细胞摄取的制造方法与工艺
- 一种鸭特异性CpG寡脱氧核苷酸及其应用的制造方法与工艺
- 一种IgE特异性核酸适配子、其应用及适配子药物的制造方法与工艺