含有乙内酰脲衍生物的药物组合物的制作方法
本发明涉及以具有高代谢稳定性、发挥强力的PTH样作用的乙内酰脲衍生物为有效成分的药品,提供诱导骨和/或软骨同化作用、用于促进对骨质疏松症、牙周病中的骨量减少、拔牙后的牙槽骨损伤、变形性关节病、关节软骨损伤、无动力性骨病、软骨成长不全症、低软骨形成症、骨质软化病、骨折等的预防、治疗、恢复和治愈的药物。
背景技术:
甲状旁腺激素(PTH)作用于骨、肾脏的靶细胞,作为调节钙(Ca)及磷(Pi)稳态的激素为人所知(非专利文献1)。基于PTH的对血清Ca浓度水平的维持主要通过针对消化道、骨及肾脏的直接或间接的作用而进行。PTH促进肾小管对Ca的重吸收,抑制机体内Ca向体外排泄。另外,通过提高在肾脏将维生素D转化为活性型维生素D的酶的合成,从而有助于促进基于活性型维生素D从消化道的Ca吸收。另外,PTH通过成骨细胞等间接促进破骨细胞的分化,从而促进从骨中释出Ca。认为上述PTH的作用主要通过基于腺苷酸环化酶及/或磷脂酶C的受体的活化(基于与PTH1R结合的cAMP产生)而发挥。
对于人而言,PTH制剂[PTH(1-34)和PTH(1-84)]具有强力的骨同化作用,诱发骨密度和骨强度的显著上升。现在,能为人利用的骨质疏松症治疗药大多是骨吸收抑制药,积极地提升骨密度之类的具有骨同化作用的药物仅有PTH制剂。因此,虽然认为PTH制剂是骨质疏松症治疗最有效手段之一(非专利文献2),但由于其是肽,所以需要侵入给予。因此,期待创造出具有PTH样作用、并且能非侵入地给予的药剂。
变形性关节病是具有下述特征的变性性疾病:膝、股关节、脊椎、手指等全身关节的软骨的变性、破坏、滑膜炎、软骨下骨的硬化、骨刺形成和慢性疼痛而带来的关节功能不全,65岁以上的人口的40%以上会患病,在医疗经济学上成为大的负担(非专利文献3、非专利文献4)。变形性关节病的原因可列举对关节软骨的物理性的过分加重、滑膜和骨髄的炎症、软骨基质成分的遗传因素、软骨下骨的骨代谢亢进等,抑制关节软骨的变性、破坏的治疗药尚未市,医疗上的需求依然高。
目前,作为治疗药的靶标,与软骨基质的破坏相关的蛋白聚糖酶(ADAMTS-4,ADAMTS-5等)、基质金属蛋白酶(MMP-3,MMP-9,MMP-13等,非专利文献5)、炎症性细胞因子(IL-1,IL-6等,非专利文献6)受到关注,但尚未被实用化。另一方面,以软骨下骨的代谢更新亢进作为靶标的药剂(利塞膦酸盐,降钙素,非专利文献7、非专利文献8)的临床试验也在进行,但未能抑制关节软骨的变性、破坏。另外,在除该机制外还兼具骨形成促进作用和软骨形成促进作用的雷奈酸锶的临床试验中,显示出了抑制关节软骨的破坏的效果(非专利文献9),但尚未被实用化。
但是,通过近年的研究,对于变形性关节病的发病机理,报告有关节软骨由永久软骨形式转化为钙化软骨,它的抑制作为治疗药的靶标受到关注(非专利文献10)。基于该作用机理,报告有抑制关节软骨的最终分化的多个机理的药剂在变形性关节病的模型动物中抑制关节软骨的变性、破坏,暗示基于该机理的治疗药的实用化的可能性(非专利文献11、非专利文献12)。
在上述情况下,本申请人发现,下述通式(A)所示的化合物或其药理学上可接受的盐作为具有PTH样作用的化合物(优选PTH1R激动剂)有用,作为骨质疏松症、骨折、骨质软化病、关节炎、血小板减少症、甲状旁腺功能减退、高磷血症或肿瘤状钙质沉着症等的予防及/或治疗、或干细胞动员有用,此前进行了专利申请(专利文献1)。
〔式中,W、X、Y、m、n、R1、R2、R33、R34参照专利文献1〕。
但是,对于在临床上价值高的能非侵入地给予的药剂的创造而言,需要考虑针对靶标的直接作用、药物的吸收、分布、代谢、排泄等体内动态。因此,为了能经口给予,期待介由人PTH1R的cAMP产生能力强、并且具有相对于人肝微粒的代谢稳定性高的PTH样作用的药剂。
现有技术文献
专利文献
专利文献1:国际公开第2010/126030号
非专利文献
非专利文献1:Kronenberg,H.M.,et al.,In Handbook of Experimental Pharmacology,Mundy,G.R.,and Martin,T.J.,(eds),pp.185-201,Springer-Verlag,Heidelberg(1993)
非专利文献2:Tashjian and Gagel,J.Bone Miner.Res.21:354-365(2006)
非专利文献3:Sem Arth Rheumatism 2013;43:303-13
非专利文献4:CPMP/EWP/784/97Rev.1.2010,European Medicines Agency
非专利文献5:Osteoarth Cart 2010;18:1109-1116
非专利文献6:Osteoarth Cart 2013;21:16-21
非专利文献7:Arthritis Rheum.2006;54(11):3494-507
非专利文献8:J Clin Pharmacol.2011;51(4):460-71
非专利文献9:Ann Rheum Dis.2013Feb;72(2):179-86
非专利文献10:Arth Rheum 2006;54(8):2462-2470
非专利文献11:Nat Med 2009;15(12):1421-1426
非专利文献12:Sci Trans Med 2011;3:101ra93。
技术实现要素:
发明要解决的问题
本发明提供通过具有高代谢稳定性、发挥强力的PTH样作用的乙内酰脲衍生物非侵入地全身暴露或局部暴露而诱导骨/软骨同化作用,促进对骨质疏松症、牙周病中的骨量减少、拔牙后的牙槽骨损伤、变形性关节病、关节软骨损伤、无动力性骨病、软骨成长不全症、低软骨形成症、骨质软化病、骨折等的预防、治疗、恢复和治愈的方法。
解决问题的方法
在上述情况下,本发明人反复进行了研究,结果发现,新发现的本发明的乙内酰脲衍生物在强制表达了人PTH1R的细胞中显现强cAMP产生能力,并且,相对于人肝微粒的代谢具有高稳定性。另外,本发明人发现通过给予本发明的化合物,诱导骨/软骨同化作用,作为用于促进对骨质疏松症、牙周病中的骨量减少、拔牙后的牙槽骨损伤、变形性关节病、关节软骨损伤、无动力性骨病、软骨成长不全症、低软骨形成症、骨质软化病、骨折等的预防、治疗、恢复和治疗的药物组合物有用。
即,本发明涉及以下方案。
〔1〕药物组合物,其用于诱导骨和/或软骨同化作用,所述药物组合物含有下述通式(1)所示的化合物或其药理学上可接受的盐作为有效成分;
〔式中,
R1及R2在不是R1和R2均为氢原子的条件下,各自独立地为:
1)氢原子、
2)卤原子、
3)可以被1~5个氟原子取代的碳数1~2个的烷基、或
4)可以被1~5个氟原子取代的碳数1~2个的烷氧基;或者,
R1及R2为相互键合而形成的下式表示的基团:
(式中的各*表示与苯基部分的键合部位。)
并且,R3及R4各自独立地为可以被1~3个氟原子取代的甲基;或者,R3及R4与它们键合的碳原子一起形成碳数3~6的环(此处,形成环的碳原子中的一个可以被氧原子、硫原子、或可以被甲基取代的氮原子取代。)。〕
本发明的药物组合物中含有的作为有效成分的化合物,可以从上述通式(1)所示的化合物中,将R1和R2的组合为三氟甲基和氢原子、并且R3及R4与它们键合的碳原子一起形成环戊基环的化合物除外。
〔2〕〔1〕所述的药物组合物,其中,前述通式(1)所示的化合物或其药学上可接受的盐的R1及R2可从以下的组合中选择:
1)R1为氢原子或卤原子,并且R2为氢原子、三氟甲基或三氟甲氧基(其中,不包括R1和R2均为氢原子的情况);
2)R1为三氟甲基或三氟甲氧基,并且R2为氢原子或卤原子;
3)R1及R2为相互键合而形成的下式所示的基团:
(式中的各*表示与苯基部分键合的部位。)
并且,R3及R4为甲基;或者,
R3及R4与它们键合的碳原子一起形成选自以下环的环:
(式中的*表示与咪唑烷-2,4-二酮部分键合的部位。)。
〔3〕〔1〕所述的药物组合物,其中,前述通式(1)所示的化合物或其药学上可接受的盐的R1、R2从以下的组合中选择:
1)R1为三氟甲氧基,并且R2为氟原子;
2)R1为溴原子,并且R2为氢原子;
3)R1为三氟甲基,并且R2为氟原子;
4)R1为氟原子,并且R2为三氟甲氧基;
5)R1为三氟甲基,并且R2为氢原子;
6)R1为氢原子,并且R2为三氟甲氧基;
7)R1、R2为相互键合而形成的下式所示的基团:
(式中的各*表示与苯基部分键合的部位。)
并且,R3及R4为甲基;或者,
R3及R4与它们键合的碳原子一起形成选自以下环的环:
(式中的*表示与咪唑烷-2,4-二酮部分键合的部位。)。
〔4〕〔1〕所述的药物组合物,其中,前述通式(1)所示的化合物或其药学上可接受的盐的R3及R4为甲基。
〔5〕〔1〕所述的药物组合物,其中,前述通式(1)所示的化合物或其药学上可接受的盐的R3及R4与它们键合的碳原子一起形成选自以下环的环:
(式中的*表示与咪唑烷-2,4-二酮部分键合的部位。)。
〔6〕〔1〕所述的药物组合物,其中,含有选自下述组的化合物或其药理学上可接受的盐作为有效成分:
1-(4-(2-((2-(4-氟-3-(三氟甲氧基)苯基)-4-氧代(oxo)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮;
1-(4-(2-((2-(3-溴苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮:
1-(4-(2-((2-(4-氟-3-(三氟甲基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮;
1-(4-(2-((2-(3-氟-4-(三氟甲氧基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮;
1-(4-(2-((2-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮;
1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮;
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮):
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮;
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-8-甲基-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮;
5-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2-氧杂-5,7-二氮杂螺[3.4]辛烷-6,8-二酮;及
4-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-4,6-二氮杂螺[2.4]庚烷-5,7-二酮。
〔7〕〔1〕所述的药物组合物,其中,化合物为1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮或其药理学上可接受的盐。
〔8〕〔1〕所述的药物组合物,其中,化合物为1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮或其药理学上可接受的盐。
〔9〕〔1〕所述的药物组合物,其中,化合物为1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮或其药理学上可接受的盐。
〔10〕根据〔1〕所述的药物组合物,其中,药物组合物用于骨质疏松症的预防或治疗、牙周病中的骨量减少的改善、拔牙后的牙槽骨损伤的恢复促进、变形性关节病的预防或治疗、关节软骨损伤的恢复促进、无动力性骨病的预防或治疗、软骨成长不全症的预防或治疗、低软骨形成症的预防或治疗、骨质软化病的预防或治疗、或者骨折的恢复促进。
〔11〕骨和/或软骨同化作用的诱导方法,其包括将药物有效量的〔1〕~〔9〕中任一项所述的化合物或其药理学上可接受的盐给药于需要骨质疏松症的预防或治疗、牙周病中的骨量减少的改善、拔牙后的牙槽骨损伤的恢复促进、变形性关节病的预防或治疗、关节软骨损伤的恢复促进、无动力性骨病的预防或治疗、软骨成长不全症的预防或治疗、低软骨形成症的预防或治疗、骨质软化病的预防或治疗、或者骨折的恢复促进的患者。
〔12〕根据〔11〕所述的方法,其中,骨和/或软骨同化作用的诱导方法为骨质疏松症的预防或治疗方法、牙周病中的骨量减少的改善方法、拔牙后的牙槽骨损伤的恢复促进方法、变形性关节病的预防或治疗方法、关节软骨损伤的恢复促进方法、无动力性骨病的预防或治疗方法、软骨成长不全症的预防或治疗方法、低软骨形成症的预防或治疗方法、骨质软化病的预防或治疗方法、或者骨折的恢复促进。
〔13〕〔1〕~〔9〕中任一项所述的化合物或其药理学上可接受的盐在制造用于骨质疏松症的预防或治疗、牙周病中的骨量减少的改善、拔牙后的牙槽骨损伤的恢复促进、变形性关节病的预防或治疗、关节软骨损伤的恢复促进、无动力性骨病的预防或治疗、软骨成长不全症的预防或治疗、低软骨形成症的预防或治疗、骨质软化病的预防或治疗、或者骨折的恢复促进的药物组合物中的应用。
〔14〕〔1〕~〔9〕中任一项所述的化合物或其药理学上可接受的盐在制造用于诱导骨和/或软骨同化作用的药物组合物中的应用。
〔15〕〔1〕~〔9〕中任一项所述的化合物或其药理学上可接受的盐,其用于骨质疏松症的预防或治疗、牙周病中的骨量减少的改善、拔牙后的牙槽骨损伤的恢复促进、变形性关节病的预防或治疗、关节软骨损伤的恢复促进、无动力性骨病的预防或治疗、软骨成长不全症的预防或治疗、低软骨形成症的预防或治疗、骨质软化病的预防或治疗、或者骨折的恢复促进。
另外,本发明提供可通过骨和/或软骨同化作用预防、治疗和/或恢复的病症的处置方法,所述方法通过给予通式(1)或其药学上可接受的盐而进行。
发明的效果
通过本发明,可提供通过使用具有强PTH样作用、并且具有高代谢稳定性的乙内酰脲衍生物,可诱导骨和/或软骨同化作用,可促进对骨质疏松症、牙周病中的骨量减少、拔牙后的牙槽骨损伤、变形性关节病、关节软骨损伤、无动力性骨病、软骨成长不全症、低软骨形成症、骨质软化病、骨折等的预防、治疗、恢复和/或治愈。
附图说明
[图1]显示对摘除卵巢的大鼠反复给药6周下的腰椎和大腿骨的骨密度的图。即,显示使用双重X射线骨盐量测定装置测定对摘除卵巢的大鼠一天一次反复给予媒介、化合物7或hPTH(1-34)6周的情况下的腰椎和大腿骨的骨密度的结果的图。
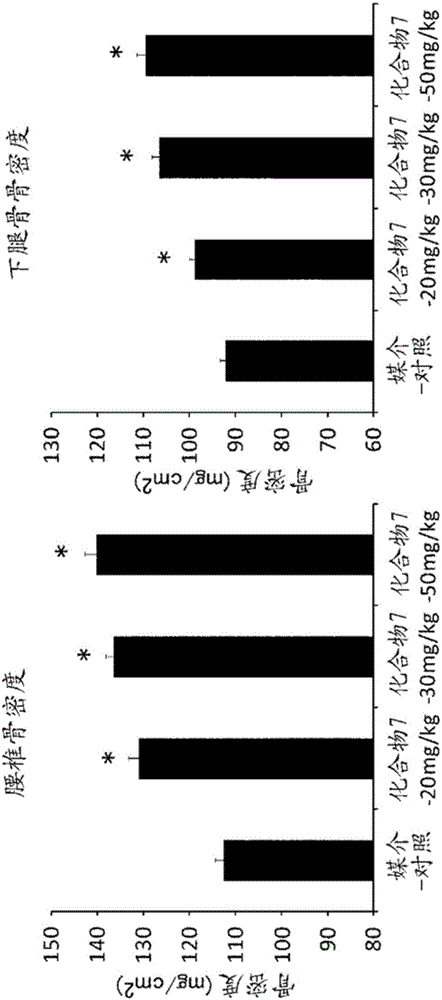
[图2]显示对正常大鼠反复给药4周下的腰椎和下腿骨的骨密度的图。即,显示使用双重X射线骨盐量测定装置测定对正常大鼠一天一次反复给予媒介、化合物7或hPTH(1-34)4周的情况下的腰椎和下腿骨的骨密度的结果的图。
[图3]显示对正常大鼠反复给药4周下的下颚骨的骨密度的图。即,显示使用双重X射线骨盐量测定装置测定对正常大鼠一天一次反复给予媒介、化合物7或hPTH(1-34)4周的情况下的下颚骨的骨密度的结果的图。
[图4]显示化合物7对兔下腿骨关节软骨细胞的最终分化的抑制作用的图。即,显示利用碱性磷酸酶染色(A)、茜素红S染色(B)评价化合物7及hPTH(1-34)对兔下腿骨关节软骨细胞的最终分化的抑制作用的结果的图。
[图5]显示人软骨细胞中的蛋白多糖合成量的图。即,显示评价化合物7及hPTH(1-34)对人软骨细胞中的蛋白多糖合成的促进作用的结果的图。
[图6]显示兔半月板部分切除模型的下腿骨关节软骨的病变部分面积比例的图。即,显示测定对兔半月板部分切除模型的关节内持续给予媒介或化合物7的情况下的下腿骨关节软骨的病变部分面积比例的结果的图。
[图7]显示兔半月板部分切除模型的下腿骨关节软骨的手术后2周时的肉眼的变化的图。即,显示肉眼观察对兔半月板部分切除模型的关节内持续给予媒介或化合物7的情况下的手术后2周时的下腿骨关节软骨的变化的结果的图。
[图8]显示对正常大鼠反复经口给药4周后的代表例的大腿骨远端关节软骨的组织画像的图。即,显示对正常大鼠反复经口给予媒介或化合物7四周后,利用光学显微镜病理组织学观察代表例的大腿骨远端关节软骨的结果的图。
[图9]为表示以30mg/kg的用量向TPTX大鼠模型经口给予时的、直到给予各化合物后24小时为止的血清Ca浓度平均变化量的图。
[图10]为显示使用双重X射线骨盐量测定装置测定对成熟龄的摘除卵巢的大鼠一天一次反复给予媒介、化合物7或hPTH(1-34)3个月的情况下的腰椎和大腿骨的骨密度的结果的图。
[图11]显示针对向兔半月板部分切除模型的关节内给予媒介或化合物7的、4周后的膝关节软骨变化进行组织学评价的结果的图。
具体实施方式
本发明涉及具有高代谢稳定性且发挥强PTH样作用的乙内酰脲衍生物及其利用。本发明人合成了上述式(1)所示的化合物或其药理学上可接受的盐,并发现了该化合物或其盐诱导骨和/或软骨同化作用。
本说明书中的“烷基”是从脂肪族烃除去1个任意的氢原子而衍生出的1价的基团,包括在骨架中不含杂原子或不饱和碳-碳键、含有氢及碳原子的烃基或烃基结构的部分集合。烷基包括直链状及支链状的结构。作为烷基,优选为碳原子数1或2的烷基。作为烷基,具体而言,可举出甲基、乙基,优选为甲基。
本说明书中的“烷氧基”是指键合有上述定义的“烷基”的氧基,优选为碳原子数1或2的烷氧基。具体而言,可举出例如甲氧基、乙氧基,优选为甲氧基。
本说明书中,“可以被A取代的B”表示B中的任意的氢原子可以被任意数量的A取代。
另外,本发明中,对于取代基的数目没有特别限制,可举出例如取代基的数目为1~5个、1~4个、1~3个、1~2个或1个等的情况。
本说明书中的“卤原子”是指氟原子、氯原子、溴原子或碘原子。
本说明书中,化学式中的“*”是指键合部位。
本发明的上述式(1)所示的化合物具有强PTH样作用,并且具有高代谢稳定性。
本说明书中的“PTH样作用”是指,作用于PTH受体,或作用于经由PTH受体的信号传递系统,产生细胞内cAMP(cAMP:环状腺苷一磷酸)的活性。
本发明中,“强PTH样作用”、“PTH样作用强”或“具有强力的PTH样作用”是否存在,例如可通过按照J.Bone.Miner.Res.14:11-20,1999中记载的方法、进行cAMP信号传递分析、测定cAMP信号活性来确认。具体而言,例如,按照参考试验例1中记载的方法,使用市售的cAMP EIA试剂盒(例如,Biotrack cAMP EIA system,GE health care),针对强制表达人PTH1R的细胞中的cAMP产生量,以给予100nM的人PTH(1-34)时的cAMP信号活性作为100%,测定各化合物显示20%cAMP信号活性的浓度(EC20)或50%cAMP信号活性的浓度(EC50)。本发明中的“强PTH样作用”或“PTH样作用强”例如是指,通过上述的方法测定的EC20的值(μM)优选为5.0以下,更优选为3.0以下,进一步优选为2.0以下。在EC50的情况下,例如,通过上述的方法测定的值(μM)优选为25.0以下,更优选为15.0以下,进一步优选为10.0以下。
另外,“高代谢稳定性”或“代谢稳定性高”是否存在,可使用常规测定方法确认。例如可使用肝细胞、小肠细胞、肝微粒、小肠微粒体、肝S9等进行确认。具体而言,例如,可通过按照T Kronbach等人的文献(Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4.Mol.Pharmacol,1989,36(1),89-96)的记载测定肝微粒中的化合物的稳定性而进行确认。更具体而言,可按照参考试验例3中记载的方法进行确认。本发明的“高代谢稳定性”或“代谢稳定性高”例如是指,上述参考试验例中记载的使用了人肝微粒的代谢稳定性试验中的清除率(μL/min/mg)的值优选为60以下,更优选为40以下,进一步优选为35以下。尤其是,在上述式(1)中,将R1与R2的组合为三氟甲基与氢原子、并且R3和R4与它们键合的碳原子一起环戊基环的情况除外,可得到高代谢稳定性。
另外,"诱导骨和/或软骨同化作用"是否存在,可使用公知的方法确认。
关于骨同化作用的诱导,例如可在一定时间持续给予被测化合物后,利用常规测定方法测定骨密度或骨量,通过与对照进行比较来确认。具体而言,例如,可以根据武田的文献(Bone 2013;53(1):167-173)所记载的方法,使用双重X射线骨盐量测定装置(例如、DCS-600EX(Aloka株式会社))测定骨密度。此时,骨密度与溶剂对照相比高时,可以认为诱导了骨同化作用。本发明的化合物优选显示为与对被测对象给予例如作为骨质疏松症治疗药使用的hPTH(1-34)的临床相当用量时的骨密度增加量同等或同等以上的增加,具体而言,例如,优选骨密度相对于溶剂对照增加8~12%的情况,更优选增加12%以上的情况。
关于软骨同化作用的诱导,例如可以通过将软骨细胞在本发明的化合物存在下进行培养、测定软骨细胞的基质(例如蛋白多糖)产生量来确认。另外,也可以通过测定软骨细胞的最终分化及钙化是否收到抑制来确认。具体而言,例如可以根据关于软骨基质产生量的Loeser等的文献(Atrh Rheum 2003;48(8):2188-2196)、Ab-Rahim等的文献(Mol Cell Biochem2013;376:11-20.)所记载的方法来测定。另外,关于最终分化的抑制,可以根据Okazaki等的文献(Osteoarth Cart 2003;11(2):122-32.)的方法进行评价。此时,软骨基质产生量、最终分化及钙化与对照相比较亢进且抑制时,可以认为软骨同化作用受到诱导。作为本发明的化合物,例如优选在软骨基质产生和软骨细胞的最终分化抑制时具有与PTH同等或同等以上的效果。本发明的化合物相比PTH具有高代谢稳定性,因此对于前述病态具有充分的效果,同时可选择多个给药路径。进而,关于软骨基质产生量得到高于PTH的效果时,对于上述病态可得到比PTH优异的效果。
另外,例如,通过采取一定时间持续给予被测化合物的对象的软骨性骨,对其进行病理组织学观察,确认关节软骨和生长板的肥厚,也可以确认软骨同化作用的诱导。具体地可以在组织学上计测关节软骨和生长板的软骨的厚度。此时,软骨的肥厚与对照相比较增加时,可以认为被测化合物的软骨同化作用得到诱导。特别是软骨肥厚与PTH相比显著时,认为对前述病态有充分的效果,因此优选,更优选的是被测化合物经口给予而具有效果。
另外,例如,根据Kikuchi等的方法、(Osteoarth Cart 1996;4(2):99-110)、Sampson等的方法(Sci Transl Med 2011;3:101ra93),在向部分切除半月板而使膝关节不稳定化而诱导变形性关节病的动物(啮齿类和非啮齿类)一定时间持续给予被测化合物后,对该膝关节部位的关节软骨的变性状态进行肉眼或病理组织学评价,由此也可以确认。此时与PTH同样,若该膝关节的关节软骨的变性得到抑制,则可以判断具有基于被测化合物的软骨同化作用和最终分化抑制作用的效果。更优选的是这些效果是通过被测化合物的经口给予而得到的。
另外,例如,根据Wakitani等的方法(Bone Joint Surg Br.1989;71(1):74-80.),将被测化合物向关节软骨和软骨下骨损伤的对象给予一定时间,通过分析损伤部的软骨的再生状況也可评价。此时,若相对于对照可观察到优异的软骨再生效果,则可以认为被化合物的软骨同化作用得到诱导。特别是,软骨再生效果相比于PTH显著时,认为对于前述病态有充分的效果,因此优选,更优选的是被测化合物经口给予而发挥效果。
这些效果可以通过测定PTH样作用进行确认。将PTH的受体即PTH1R通过旁分泌而活性化的PTHrP,是与软骨细胞的增殖和分化调节相关的重要因子,已知有抑制软骨细胞的最终分化、维持软骨组织的作用(Science 1996;273:663-666.)。基于PTH1R的活性化的软骨同化作用可通过根据例如Xie等的方法(Human Mol Genet 2012;21(18):3941-3955)、向正常或具有遗传性生长障碍的对象一定时间给予被测化合物,分析软骨性骨的生长速度和组织学的生长板的肥厚,由此进行评价。此时,若相比于对照可确认到生长速度、生长板的肥厚,则可判断具有被测化合物的软骨同化作用。特别是优选被测化合物的效果比PTH优异,更优选的是被测化合物经口给予而具有效果。
本发明涉及的化合物可以是游离体,但药理学上可接受的盐也包含在本发明中。作为这样的“盐”,可举出例如无机酸盐、有机酸盐、无机碱盐、有机碱盐、酸性或碱性氨基酸盐等。
作为无机酸盐的优选例,可举出例如盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、磷酸盐等,作为有机酸盐的优选例,可举出例如乙酸盐、琥珀酸盐、富马酸盐、马来酸盐、酒石酸盐、柠檬酸盐、乳酸盐、硬脂酸盐、苯甲酸盐、甲磺酸盐、苯磺酸盐、对甲苯磺酸盐等。
作为无机碱盐的优选例,可举出例如钠盐、钾盐等碱金属盐、钙盐、镁盐等碱土金属盐、铝盐、铵盐等,作为有机碱盐的优选例,可举出例如二乙基胺盐、二乙醇胺盐、葡甲胺盐、N,N-二苄基乙二胺盐等。
作为酸性氨基酸盐的优选例,可举出例如天冬氨酸盐、谷氨酸盐等,作为碱性氨基酸盐的优选例,可举出例如精氨酸盐、赖氨酸盐、鸟氨酸盐等。
通过将本发明的化合物放置于大气中,有时吸收水分,带有吸附水,或形成水合物,这样的水合物也属于本发明的盐。
此外,本发明的化合物有时吸收其他种类的溶剂,形成溶剂合物,这样的盐也作为式(1)的化合物的盐被包含在本发明中。
本说明书中,为了方便,有时将化合物的结构式表示为一定的异构体,但本发明包括根据化合物的结构而形成的所有几何异构体、基于不对称碳的光学异构体、立体异构体、互变异构体等异构体及异构体混合物,不限于为了方便而记载的结构式,可以是任一种异构体,也可以是混合物。因此,虽然本发明的化合物中有时存在在分子内具有不对称碳原子的光学活性体及外消旋体,但本发明不受限制,包含它们中的所有。
本发明包括式(1)所示的化合物的所有同位素化合物(isotopes)。本发明化合物的同位素化合物是至少1个原子被原子序数(质子数)相同而质量数(质子和中子的数量和)不同的原子取代而成的化合物。作为本发明化合物中包含的同位素的例子,包括氢原子、碳原子、氮原子、氧原子、磷原子、硫原子、氟原子、氯原子等,分别包括2H、3H、13C、14C、15N、17O、18O、31P、32P、35S、18F、36Cl等。特别地,3H、14C这样的能呈现放射性的衰变的放射性同位素在进行药品或化合物的体内组织分布试验等时有用。稳定同位素不发生衰变,存在量几乎不发生变化,也没有放射性,因而能安全地使用。本发明的化合物的同位素化合物可通过将合成中使用的试剂替换为包含对应的同位素的试剂、按照常规方法而进行转化。
本发明涉及的化合物中,有时也存在结晶多型,但没有特别限制,可以是任一种单一晶型,也可以是晶型混合物。
本发明涉及的化合物包括其前体药物。前体药物是指本发明化合物的衍生物,其具有可化学分解或代谢分解的基团,在给予至机体后,恢复成原来的化合物,发挥本来的药效,包括非基于共价键的复合体和盐。
作为本发明的上述式(1)所示的化合物,优选如下所述。
式中,R1及R2可从以下的组合中选择:
1)R1为氢原子或卤原子,并且R2为氢原子、三氟甲基或三氟甲氧基(其中,不包括R1和R2均为氢原子的情况);
2)R1为三氟甲基或三氟甲氧基,并且R2为氢原子或卤原子;
3)R1及R2为相互键合而形成的下式所示的基团:
(式中,*分别表示与苯基部分键合的部位。)
并且,R3及R4为甲基;
或R3及R4与它们键合的碳原子一起形成选自以下环的环:
(式中的*表示与咪唑烷-2,4-二酮部分键合的部位。)。
作为本发明的上述式(1)所示的化合物,更优选如下所述。
式中,R1、R2可从以下的组合中选择:
1)R1为三氟甲氧基,并且R2为氟原子;
2)R1为溴原子,并且R2为氢原子;
3)R1为三氟甲基,并且R2为氟原子;
4)R1为氟原子,并且R2为三氟甲氧基;
5)R1为三氟甲基,并且R2为氢原子;
6)R1为氢原子,并且R2为三氟甲氧基;
7)R1、R2为相互键合而形成的下式所示的基团:
(式中,*分别表示与苯基部分键合的部位。)
并且,R3及R4为甲基;
或R3及R4与它们键合的碳原子一起形成选自以下环的环:
(式中的*表示与咪唑烷-2,4-二酮部分键合的部位。)。
作为本发明的上述式(1)所示的化合物,进一步优选为选自由以下化合物组成的组的化合物或其药理学上可接受的盐。
化合物1:1-(4-(2-((2-(4-氟-3-(三氟甲氧基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物2:1-(4-(2-((2-(3-溴苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物3:1-(4-(2-((2-(4-氟-3-(三氟甲基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物4:1-(4-(2-((2-(3-氟-4-(三氟甲氧基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物5:1-(4-(2-((2-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物6:1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物7:1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮、
化合物8:1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮、
化合物9:1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-8-甲基-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮、
化合物10:5-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2-氧杂-5,7-二氮杂螺[3.4]辛烷-6,8-二酮、及
化合物11:4-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-4,6-二氮杂螺[2.4]庚烷-5,7-二酮。
在上述化合物1~11中,更优选化合物6、7、8。
这样的本发明的上述式(1)所示的化合物或其药理学上可接受的盐诱导骨-软骨同化作用,通过该作用,对于骨质疏松症的预防或治疗、牙周病中的骨量减少的改善、拔牙后的牙槽骨损伤的恢复促进、变形性关节病的预防或治疗、关节软骨损伤的恢复促进、无动力性骨病的预防或治疗、低软骨形成症的预防或治疗、或者骨质软化病的预防或治疗有用。
本发明涉及的化合物或其盐可利用通常使用的方法制成片剂、散剂、细粒剂、颗粒剂、包衣片剂、胶囊剂、糖浆剂、含剂、吸入剂、栓剂、注射剂、软膏剂、眼软膏剂、滴眼剂、滴鼻剂、滴耳剂、巴布剂、洗剂等制剂。可使用制成制剂时通常使用的载体,例如赋型剂、结合剂、润滑剂、着色剂、矫味矫臭剂,及根据必要使用的稳定化剂、乳化剂、吸收促进剂、表面活性剂、pH调节剂、防腐剂、抗氧化剂等,可配合通常作为药品制剂的原料使用的成分,利用常规方法制成制剂。
例如,为了制造经口制剂,添加本发明涉及的化合物或其药理学上可接受的盐和赋型剂、以及根据需要使用的结合剂、崩解剂、润滑剂、着色剂、矫味矫臭剂等,然后利用常规方法,制成散剂、细粒剂、颗粒剂、片剂、包衣片剂、胶囊剂等。
作为它们的成分,可举出例如大豆油、牛脂、合成甘油酯等动植物油;液体石蜡、角鲨烷、固态石蜡等烃;肉豆蔻酸十八烷基酯、肉豆蔻酸异丙酯等酯油;十八醇十六醇混合物、山嵛醇等高级醇;硅树脂;硅油;聚氧乙烯脂肪酸酯、山梨糖醇酐脂肪酸酯、甘油脂肪酸酯、聚氧乙烯山梨糖醇酐脂肪酸酯、聚氧乙烯氢化蓖麻油、聚氧乙烯聚氧丙烯嵌段共聚物等表面活性剂;羟乙基纤维素、聚丙烯酸、羧基乙烯基聚合物、聚乙二醇、聚乙烯吡咯烷酮、甲基纤维素等水溶性高分子;乙醇、异丙醇等低级醇;甘油、丙二醇、二丙二醇、山梨糖醇等多元醇;葡萄糖、蔗糖等糖;硅酸酐、硅酸铝镁、硅酸铝等无机粉体、纯化水等。
作为赋型剂,可举出例如乳糖、玉米淀粉、白糖、葡萄糖、甘露糖醇、山梨糖醇、结晶纤维素、二氧化硅等。
作为结合剂,可举出例如聚乙烯醇、聚乙烯醚、甲基纤维素、乙基纤维素、阿拉伯胶、西黄耆胶、明胶、虫胶、羟丙基甲基纤维素、羟丙基纤维素、聚乙烯吡咯烷酮、聚丙二醇-聚氧乙烯-嵌段聚合物、葡甲胺等。
作为崩解剂,可举出例如淀粉、琼脂、明胶粉末、结晶纤维素、碳酸钙、碳酸氢钠、柠檬酸钙、糊精、果胶、羧甲基纤维素-钙等。
作为润滑剂,可举出例如硬脂酸镁、滑石、聚乙二醇、二氧化硅、氢化植物油等。
作为着色剂,可使用允许添加到药品中的着色剂,作为矫味矫臭剂,可使用可可粉、薄荷醇、芳香散、薄荷油、龙脑、肉桂粉等。
对于上述片剂、颗粒剂,当然可以包覆糖衣,此外,根据需要也可包覆其他包衣。另外,在制造糖浆剂、注射用制剂等液剂时,向本发明涉及的化合物或其药理学上可接受的盐中添加pH调节剂、溶解剂、等渗剂等,和根据需要的溶解辅助剂、稳定化剂等,利用常规方法制成制剂。
对于制造外用剂时的方法没有限制,可利用常规方法进行制造。即,作为制成制剂时使用的基剂原料,可使用药品、药物类似物(quasi drugs)、化妆品等中通常使用的各种原料。作为使用的基剂原料,具体而言,可举出例如动植物油、矿物油、酯油、蜡类、高级醇类、脂肪酸类、硅油、表面活性剂、磷脂类、醇类、多元醇类、水溶性高分子类、粘土矿物类、纯化水等原料,此外,根据需要,可添加pH调节剂、抗氧化剂、螯合剂、防腐防霉剂、着色料、香料等,本发明涉及的外用剂的基剂原料不限于这些。
另外,根据需要,也可配合具有分化诱导作用的成分、血流促进剂、杀菌剂、消炎剂、细胞活化剂、维生素类、氨基酸、保湿剂、角质溶解剂等成分。应予说明,上述基剂原料的添加量为,成为在通常制造外用剂时设定的浓度的量。
在给予本发明涉及的化合物或其盐或它们的水合物时,对其形态没有特别限定,可利用通常使用的方法经口给予或非经口给予。例如,可制成片剂、散剂、颗粒剂、胶囊剂、糖浆剂、含剂、吸入剂、栓剂、注射剂、软膏剂、眼软膏剂、滴眼剂、滴鼻剂、滴耳剂、巴布剂、洗剂等制剂而进行给予。
本发明涉及的药物的给予量和给予方法可根据症状的程度、年龄、性别、体重、给予形态、盐的种类、疾病的具体种类等适当选择。
给予量根据患者的疾病的种类、症状的程度、患者的年龄、性别差异、相对于药剂的敏感性差等的不同而显著不同,通常,作为成人,每1天给予约0.03-1000mg、优选0.1-500mg、进一步优选0.1-100mg,每1天分1次~数次给予。
给药路径考虑患者的疾病的种类、症状的程度、患者的年龄、性别差异、相对于药剂的敏感性差等而进行适当选择。给药方法没有特别限定,只要将本发明的化合物非侵入地全身暴露或局部暴露而得到骨和/或软骨同化作用的诱导效果的方法即可。作为这样的给药方法,可列举例如经口给药、静脉内给药、经鼻给药、经皮给药、经肺给药、关节内给药。
在上述式(1)所示的本发明化合物的制造中,原料化合物、各种试剂可形成盐、水合物或溶剂合物,它们均根据起始原料、使用的溶剂等的不同而不同,而且只要不抑制反应,就没有特别限制。
对于使用的溶剂,不言而喻,也根据起始原料、试剂等的不同而不同,另外,只要不抑制反应、以一定程度溶解起始物质即可,没有特别限制。
各种异构体(例如几何异构体、基于不对称碳的光学异构体、旋转异构体、立体异构体、互变异构体等)可通过使用通常的分离手段(例如重结晶、非对映异构体盐法、酶分割法、各种色谱法(例如薄层色谱法、柱色谱法、高效液相色谱法、气相色谱法等))进行纯化、分离。
当以游离体形式得到本发明涉及的化合物时,可按照常规方法转化成该化合物可以形成的盐或它们的水合物的状态。另外,当以该化合物的盐或水合物的形式得到本发明涉及的化合物时,可按照常规方法转化为该化合物的游离体。
本发明涉及的化合物的分离、纯化可应用萃取、浓缩、馏去、结晶化、过滤、重结晶、各种色谱法等通常的化学操作来进行。
本说明书中引用的所有现有技术文献作为参照并入本说明书中。
一般的合成法
本发明的化合物可利用多种方法合成。利用以下的路线说明其中一部分合成方法。路线是例举,本发明不仅限于明确记载的化学反应及条件。以下的路线中,为了说明清楚,将一部分取代基除外,但并非意在将它们限制于公开的路线。本发明的代表化合物可使用适当的中间体、公知的化合物及试剂来合成。下述一般的合成法中的式中的R1、R2、R3、R4与上述通式(1)所示的化合物(下述一般的合成法中式1所示的化合物)的R1、R2、R3、R4含义相同。
本发明的化合物(式1)可利用以下所示的制造方法(方法A、方法B)合成。
路线1(方法A)
路线1是以下方法:将羧酸衍生物(1)和氨基-酰胺衍生物(2)缩合,得到酰胺-酰胺衍生物(3),然后,利用分子内环化而构建螺咪唑酮环,得到乙内酰脲衍生物(式1)。
步骤1是使羧酸衍生物(1)与氨基-酰胺衍生物(2)反应的方法。作为偶联试剂,可举出例如N,N'-二环己基碳二亚胺(DCC)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC)、O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐(HATU)、及4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物n-水合物(DMT-MM)。作为碱,可举出三乙基胺或N,N-二异丙基乙基胺。根据需要,可使用4-(二甲基氨基)吡啶(DMAP)作为催化剂。作为适当的溶剂,可举出二氯甲烷或N,N-二甲基甲酰胺等。作为使用DMT-MM时的适当的反应溶剂,可举出甲醇、乙醇及乙腈。反应温度例如在0℃~室温进行,反应时间为0.5~24小时。对于得到的酰胺-酰胺衍生物(3),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤2是以下方法:在氢氧化钠水溶液或叔丁醇钾等适当的碱的存在下,在乙醇、叔丁醇或二甲基亚砜等适当的溶剂中,将酰胺-酰胺衍生物(3)环化。反应温度例如在室温~回流条件下进行,反应时间为1~24小时。对于得到的乙内酰脲衍生物(式1),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
路线1所示的氨基-酰胺衍生物(2)可从哌啶酮衍生物(4)合成。路线2中示出氨基-酰胺衍生物(2)的合成法。
路线2
步骤3是将哌啶酮衍生物(4)衍生为氨基-腈衍生物(5)的Strecker合成。即,在存在/不存在水的条件下,在甲醇、乙醇、或四氢呋喃等适当的溶剂中,使哌啶酮衍生物(4)与氰化钠或氰化钾、及氯化铵或乙酸铵反应的方法。反应温度例如在室温~80℃下进行,反应时间为2~72小时。对于得到的氨基-腈衍生物(5),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤4是利用在过氧化氢存在下的碱性水解条件、将腈基转化为酰胺基的方法。本反应例如可以将Chemistry-A European Journal(2002),8(2),439-450作为参考而进行。
步骤5是在H2气氛下、分别在钯碳或氢氧化钯碳等催化剂的存在下、在甲醇、乙醇、三氟乙醇、二甲基甲酰胺、或二甲基乙酰胺等惰性溶剂中、将化合物(6)的烯烃氢化的方法。反应温度在室温~80℃下进行,有时可以在加压下进行反应。对于得到的氨基-酰胺衍生物(2),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
路线2所示的哌啶酮衍生物(4)可从公知的缩酮-乙烯基磺酰基衍生物(7)和乙内酰脲-芳基溴化物衍生物(8)合成。路线3中示出哌啶酮衍生物(4)的合成法。
路线3
步骤6是以下方法:在N2气氛下,分别地,在三(二苄叉丙酮)二钯(0)、双(二苄叉丙酮)钯等钯催化剂的存在下,添加四氟硼酸三叔丁基膦等膦配体、甲基二环己基胺等适当的碱,在N-甲基-2-哌啶酮(NMP)等适当的溶剂中,将缩酮-乙烯基磺酰基衍生物(7)和乙内酰脲-芳基溴化物衍生物(8)偶联,由此合成缩酮-芳基乙烯基磺酰基衍生物(9)。反应温度在90℃~回流温度下进行。对于得到的缩酮-芳基乙烯基磺酰基衍生物(9),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤7是以下方法:针对缩酮-芳基乙烯基磺酰基衍生物(9),在含水四氢呋喃等适当的溶剂中,在盐酸等酸的存在下,将缩酮转化为酮。反应温度例如为溶剂的沸点,反应时间为1~24小时。对于得到的哌啶酮衍生物(4),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
路线3所示的乙内酰脲-芳基溴化物衍生物(8)可由4-溴-3,5-二甲基苯胺(10)和溴乙酸衍生物(11)或2-溴-5-碘-1,3-二甲基苯(13)和氨基酸衍生物(14)合成。路线4中示出乙内酰脲-芳基溴化物衍生物(8)的合成法。
路线4
步骤8是以下方法:在二异丙基乙基胺等适当的碱存在下,在N-甲基-2-哌啶酮(NMP)等适当的溶剂中,用溴乙酸衍生物(11)将4-溴-3,5-二甲基苯胺(10)烷基化的方法。反应温度例如为室温~100℃,反应时间为1~24小时。对于得到的芳基溴化物-氨基酸衍生物(12),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤9是以下方法:在碘化铜(I)等金属催化剂的存在下,使2-溴-5-碘-1,3-二甲基苯(13)与氨基酸衍生物(14)反应,合成芳基溴化物-氨基酸衍生物(12)。反应可按照以下方式进行:在二氮杂双环十一碳烯(DBU)等适当的碱存在下,在N,N-二甲基乙酰胺(DMA)等适当的溶剂中,在80-120℃左右的反应温度下进行。对于得到的芳基溴化物-氨基酸衍生物(12),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤10是以下方法:在酸性条件下,使芳基溴化物-氨基酸衍生物(12)与氰酸钠反应,合成乙内酰脲-芳基溴化物衍生物(8)。关于溶剂,例如用乙酸-二氯甲烷等混合溶剂进行,反应温度为室温~60℃,反应时间为1~24小时。对于得到的乙内酰脲-芳基溴化物衍生物(8),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
路线3所示的乙内酰脲-芳基溴化物衍生物(8)也可从4-溴-3,5-二甲基苯胺(10)和酮衍生物(15)合成。路线5示出乙内酰脲-芳基溴化物衍生物(8)的合成法。
路线5
步骤11是将酮衍生物(15)衍生为芳基氨基-腈衍生物(16)的Strecker合成。即,是以下方法:针对酮衍生物(15),在乙酸等适当的溶剂中,使4-溴-3,5-二甲基苯胺(10)与三甲基氰硅烷反应。可在室温等反应温度下进行,反应时间为1~3小时左右。对于得到的芳基氨基-腈衍生物(16),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤12是以下方法:在二氯甲烷等适当的溶剂中,使芳基氨基-腈衍生物(16)与2,2,2-三氯乙酰基异氰酸酯反应,然后添加甲醇、水、三乙基胺等试剂,在加热条件下进行反应,合成亚氨基乙内酰脲衍生物(17)。对于得到的亚氨基乙内酰脲衍生物(17),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤13是以下方法:在酸性条件下,将亚氨基乙内酰脲衍生物(17)转化为乙内酰脲-芳基溴化物衍生物(8)。例如,可在乙酸-水溶剂中,在65℃左右的加热条件下,以1~6小时左右的反应时间进行合成。对于得到的乙内酰脲-芳基溴化物衍生物(8),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
路线6是以下方法:在金属催化剂存在下,使乙烯基磺酰胺衍生物(18)与乙内酰脲-芳基溴化物衍生物(8)进行Heck反应,然后对化合物(19)的烯烃进行催化氢化,得到乙内酰脲衍生物(式1)。
路线6(方法B)
步骤14的反应可按照步骤6的方法,步骤15的反应可按照步骤5的方法,合成乙内酰脲衍生物(式1)。对于得到的乙内酰脲衍生物(式1),利用常规技术进行分离,根据需要,可利用结晶化或色谱法进行纯化。
步骤14中使用的乙烯基磺酰胺衍生物(18)可将WO2010/126030(A1)的路线2、路线3、及路线12作为参考而进行合成。
应予说明,本说明书中引用的所有现有技术文献,作为参照并入本说明书中。
本发明通过以下的实施例进一步例示,但不受下述实施例限定。
实施例
实施例1:对于向摘除卵巢的大鼠反复给药6周的骨密度的效果
将通过日本Charles River株式会社购入的雌性Crl:CD(SD)大鼠在20~26℃、湿度35~75%的标准试验室条件下驯化1周以上后,用于实验。对于大鼠,自由摄取自来水以及包含1.1%钙、1.0%磷酸和250IU/100g的维生素D3的标准啮齿动物饲料(CE-2)(日本CLEA株式会社)。
对12周龄的大鼠实施两侧的卵巣摘除术(OVX)及假手术(Sham)。在手术后第4周测定体重,1组6只将大鼠分配进行分组,以使各组的平均体重达到均等。分组的第二天起对各大鼠1天1次反复给药6周。Sham-对照组的大鼠分别经口和皮下给予需经口给予的溶剂(媒介)及皮下给予的溶剂(PC buffer)。OVX-对照组的大鼠分别经口和皮下给予媒介及PC buffer。OVX-化合物7的大鼠以30mg/kg的用量经口给予溶于媒介的上述化合物7,或者皮下给予PC buffer。OVX-hPTH(1-34)组的大鼠经口给予媒介,以0.9nmol/kg的用量皮下给予溶于PC buffer的hPTH(1-34)。
此外,以与对人给予作为骨质疏松症治疗药而在临床应用的Forteo(注册商标)20μg时相同程度的AUC(血中浓度―时间曲线下面积)的方式,将对大鼠给药时所显示的给药用量设定为0.9nmol/kg。每组均分别经口给予5mL/kg的用量、皮下给予1mL/kg的用量。媒介使用将10%二甲基亚砜(和光纯药工业株式会社)、10%KolliphorEL(Sigma-Aldrich Japan合同会社)、10%羟基丙基‐β‐环糊精(日本食品化工株式会社)利用甘氨酸(和光纯药工业株式会社)及氢氧化钠(和光纯药工业株式会社)制备为pH10而得的组成。PC buffer使用将25mmol/L磷酸-枸橼酸缓冲液、100mmol/L NaCl、0.05%Tween80制备为pH5.0而得的组成。在最终给药的第二天在麻醉下通过腹大动脉采取血液而将大鼠安乐死处置后,进行剖检采取腰椎及大腿骨。腰椎及大腿骨保存于70%乙醇中。使用双重X射线骨盐量测定装置(DCS-600EX、Aloka株式会社)测定腰椎及大腿骨的骨密度。腰椎骨密度测定第2至第4腰椎,大腿骨骨密度测定是将大腿骨纵向10等分、测定膝侧远端的3份。将结果示于图1。
数据以平均值+标准偏差(SE)显示。使用SAS前临床手册ver.5.00(SAS Institute Japan),实施以下的统计学解析。显著性水平为两侧5%。关于腰椎及大腿骨骨密度,通过2组的t检测进行Sham-对照组和OVX-对照组的比较(#P<0.05)、OVX-对照组和OVX-化合物7组的比较(*P<0.05)、及OVX-对照组和OVX-hPTH(1-34)组的比较(∫P<0.05)。
如图1所示,在大腿骨骨密度中,OVX-对照组相对于Sham-对照组显示出显著的骨密度减少,作为阳性对照的OVX-hPTH(1-34)组相对于OVX-对照组显示出显著的增加。OVX-hPTH(1-34)组相对于OVX-对照组的增加率为8%。此时OVX-化合物7组相对于OVX-对照组显示出显著的增加,增加率为12%。另外,在腰椎骨密度中,OVX-对照组相对于Sham-对照组显示出显著的骨密度减少,OVX-化合物7组相对于OVX-对照组不显著,但显示出增加倾向。OVX-hPTH(1-34)组相对于OVX-对照组显示出显著的增加,增加率为12%。
如上所述化合物7的反复经口给予时,骨质疏松症的病态模型即OVX大鼠的骨密度增加,因此认为化合物7对骨质疏松症、牙周病中的骨量减少、抜牙后的牙槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨再建的疾病的预防、治疗、改善、恢复促进有效。进而,对于通式(1)所示的化合物,在参考试验例1至5中可确认强的PTH样作用和高代谢稳定性,认为通过基于PTH样作用的骨同化作用可得到骨密度增加作用。认为关于通式(1)所示的化合物,对骨质疏松症、牙周病中的骨量减少、抜牙后的牙槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨再建的疾病的预防、治疗、改善、恢复促进有效。
实施例2:对于对正常大鼠反复给药4周的骨密度的效果
将从株式会社日本医科学动物資材研究所购入的雌性RccHan:WIST大鼠在20~26℃、湿度30~70%的标准实验室条件下驯化1周以上后,用于实验。对于大鼠,自由摄取自来水以及标准啮齿动物饲料(CR-LPF)(Oriental酵母工业株式会社)。
对8周龄的大鼠留置静脉导管。导管从鼠径部的大腿静脉插入,前端延伸至后大静脉而留置。在术后1周测定体重,1组10只将大鼠分配分组,以使各组的平均体重达到均等。分组的2天后将全部大鼠1天1次反复静脉内给药4周。将留置的导管连接输液泵(MEDFUSION SYRINGEINFUSION PUMP Model 2001)进行给药。
媒介-对照组静脉内给予溶剂(媒介)。化合物7―20mg/kg、30mg/kg、50mg/kg组分别以20mg/kg、30mg/kg、50mg/kg的用量静脉内给予溶于媒介的化合物7。每组的给予容量均为5mL/kg、给予速度为5mL/kg/分钟。媒介使用5%二甲基亚砜(和光纯药工业株式会社)、25%丙二醇(关东化学株式会社)/20%乙醇(纯正化学株式会社)/15%羟基丙基‐β‐环糊精(日本食品化工株式会社)/300mM甘氨酸(和光纯药工业株式会社)/192mM氢氧化钠(和光纯药工业株式会社)/生理食盐水(株式会社大塚制药工厂)的组成。最终给药的第二天在麻醉下通过腹大动脉采取血液对大鼠进行安乐死处置后,进行剖检采取腰椎、下腿骨、及下颚骨。腰椎、下腿骨、及下颚骨保存于70%乙醇中。腰椎(第2至第4腰椎)、下腿骨、及下颚骨的骨密度使用双重X射线骨盐量测定装置(DCS-600EX、Aloka株式会社)测定。结果示于图2、图3。
数据以平均值+标准偏差(SE)显示。使用SAS前临床手册ver.5.00(SAS Institute Japan),实施以下的统计学解析。显著性水平为两侧5%。关于腰椎、下腿骨、及下颚骨骨密度,以媒介-对照组为对照组,进行相对于化合物7的3用量组的参数Dunnett型多重比较(*P<0.05)。
如图2所示,在腰椎及下腿骨骨密度中,化合物7给予组相对于媒介-对照组在用量依存性上显示出显著的骨密度增加效果。另外,化合物7―20mg/kg组、30mg/kg组、50mg/kg组3组的相对于媒介-对照组的腰椎骨密度的增加率分别为16%、21%、25%,下腿骨骨密度的增加率为7%、16%、19%。另外,如图3所示,下颚骨骨密度中,化合物7-30mg/kg组相对于媒介-对照组显示出显著的骨密度增加效果。相对于媒介-对照组的增加率为7%。
如上所述化合物7的反复经口给予时,骨质疏松症的病态模型即OVX大鼠的大腿骨骨密度增加,化合物7的反复静脉内给予时,正常大鼠的腰椎、下腿骨、及下颚骨骨密度增加,因此认为化合物7的全身暴露对骨质疏松症、牙周病中的骨量减少、抜牙后的压槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨的疾病的预防、治疗、改善、恢复促进有效。进而,对于通式(1)所示的化合物,在参考试验例1至5中可确认强的PTH样作用和高代谢稳定性,认为通过基于PTH样作用的骨同化作用可得到骨密度增加作用。因此,认为关于通式(1)所示的化合物,对骨质疏松症、牙周病中的骨量减少、抜牙后的牙槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨再建的疾病的预防、治疗、改善、恢复促进有效。
实施例3:化合物7对于兔关节软骨细胞的最终分化的抑制效果
将NZW系兔(4周龄、Oriental酵母工业株式会社)安乐死处置后,采取下腿骨的关节软骨,转移至50mL试验管(日本Becton Dickinson株式会社)。向其中添加含1%胰蛋白酶(和光纯药工业株式会社)的PBS(Nacalai Tesque株式会社),在37℃消化软组织1小时。然后1,200rpm,5分钟离心分离后除去上清,加入PBS(-),悬浮软骨组织,1,200rpm,5分钟离心分离后,除去上清,用PBS(-)悬浮洗涤,重复3次。1,200rpm,5分钟离心分离后,在细胞板中用含0.2%Type II collagenase(CLS-2,Worthington Biochemical Corp.)的DMEM(Life Technologies Japan株式会社)在37℃下消化3小时,添加胎牛血清(Life Technologies Japan株式会社)10%v/v,停止反应,用10mL移液管(日本Becton Dickinson株式会社)强力移液分离软骨细胞。然后,1,200rpm,5分钟离心分离,除去上清,用含10%FCS的DMEM悬浮洗涤,重复3次,将软骨细胞以1x104个/孔接种于覆盖了I型胶原的的96孔培养板(AGC TECHNO GLASS CO.,LTD.,)。1周3次更换培养基,细胞达到融合后用含100μg/mL磷酸抗坏血酸镁盐n-水合物(和光纯药工业株式会社)、10mmol/Lβ甘油磷酸5水合物(和光纯药工业株式会社)、10%FCS的DMEM培养。此时,按照以下条件培养,进行碱性磷酸酶染色(培养14天)和茜素红染色(培养21天),评价软骨细胞的最终分化。
1)对照
2)BMP-2 100ng/mL
3)化合物7 10-7mol/L
4)化合物7 10-6mol/L
5)化合物7 3×10-6mol/L
6)化合物7 10-5mol/L
7)PTH(1-34) 10-10mol/L
8)PTH(1-34) 10-9mol/L
9)PTH(1-34) 10-8mol/L
10)PTH(1-34) 10-7mol/L。
关于碱性磷酸酶染色,废弃培养基后,将软骨细胞用200mmol/L Tris-HCl pH8.2缓冲液洗涤1次,按照碱性磷酸酶染色试剂(Vector Red Alkaline Phosphatase Substrate Kit I,Vector Laboratories,Inc.)的操作步骤进行染色,用倒立显微鏡(株式会社Nikon)拍照(4倍目标)。
其结果,BMP-2显示出碱性磷酸酶活性的增加。化合物7、PTH(1-34)均在浓度依存地抑制碱性磷酸酶活性(图4A)。
关于茜素红染色,废弃培养基后,用PBS洗涤软骨细胞2次,用100%乙醇(和光纯药工业株式会社)固定15分钟,废弃乙醇后用1%茜素红S(和光纯药工业株式会社)染色15分钟后,用蒸馏水洗涤,用倒立显微鏡拍照。其结果,BMP-2的茜素红染色性显著地增加,促进钙化。化合物7、PTH(1-34)均浓度依存地抑制茜素红染色性,抑制钙化(图4B)。
实施例4:化合物7对于人关节软骨细胞蛋白多糖合成的效果
购入冷冻保存的人关节软骨细胞(Lot 2867,Cell Applications Inc.,)后,在37℃水浴中解冻,向T75培养烧瓶(CORNING,Corning Japan K.K.)加入含10%Growth Supplement的Basal Medium(Growth Medium,Cell Applications Inc.)15mL进行培养。第二天,用Growth Medium 15mL交换,培养3天。除去Growth Medium,用HBSS(Cell Applications Inc.)洗涤软骨细胞层,除去。加入1mL的胰蛋白酶/EDTA溶液(Cell Applications Inc.),在室温下静置约5分钟,将软骨细胞从烧瓶剥离。加入10mL的Neutalizing solution(Cell Applications Inc.),使用Bulker-Turk血球计算板,计测细胞数后,移至15mL试验管,离心分离(1,200rpm,5分钟、株式会社TOMY SEIKO),制成软骨细胞板。丢弃上清,以1.2%海藻酸钠(25mmol/L HEPES/150mmol/L氯化钠溶液,pH7.0)中达到2×106个/mL的方式进行制备,吸引至带22G注射针头的1mL注射器(TERUMO株式会社),向加入了102mmol/L CaCl2水溶液2mL的24孔板(Corning Japan K.K.)的孔中以每孔5滴滴下,静置5分钟,形成小珠(beads)。然后,用150mmol/L氯化钠溶液洗涤3次,用Growth Medium培养1天,交换至含1%Growth supplement的Basal Medium(Cell Applications Inc.)。此时,向培养基添加以下因子,1周3次交换培养基,培养13天。
1)对照
2)TGF-β1 10ng/m
3)PTH(1-34) 10-8mol/L
4)化合物7 10-6mol/L
5)化合物7 10-5mol/L。
培养12天添加35S标记硫酸(株式会社PerkinElmer Japan),以达到370kBq/孔,24小时后将培养基回收至试验管中,在4℃保存。向海藻酸凝胶中添加55mmol/L枸橼酸钠溶液(Nacalai Tesque株式会社、1mL/孔),在37℃下培养10分钟,使其溶胶化,回收至微管(Eppendorf株式会社)中,离心分离(1,200rpm,5分钟),制成软骨细胞板。将微管用含1mg/mL actinase E(科研制药株式会社)的0.2mol/L Tris-HCl(Sigma-Aldrich Co.LLC.)/5mmol/L CaCl2(Nacalai Tesque株式会社)pH7.8 0.5mL悬浮,移至12孔板(Corning Japan K.K.),密封,在设定为50℃的孵育器(Espec株式会社)中培养一晩。将该消化液0.4mL移至试验管,添加0.1mg/mL软骨素硫酸(和光纯药工业株式会社)水溶液250μL、2mmol/L MgSO4(和光纯药工业)、0.2mol/L Tris-HCl(Sigma-Aldrich)/5mmol/L CaCl2(Nacalai Tesque株式会社)pH7.8)、1%Cetylpyridinium chrolide(CPC,和光纯药工业株式会社)/20mmol/L NaCl(Nacalai Tesque株式会社)各2.5mL,在37℃下培养3小时。将该溶液在真空泵吸引下用玻璃滤器(GC-50,ADVANTEC)过滤,用1%CPC/20mmol/L NaCl洗涤,除去游离的35S标记硫酸。将滤器移至液体闪烁计数器用的瓶中,加入5mL的闪烁体(Hionic-Fluor,株式会社PerkinElmer Japan)用液体闪烁计数器(TRI-CARB,株式会社PerkinElmer Japan)测定放射活性。
消化液的剩余的0.1mL用于DNA定量。向DNA定量试剂(Cosmo Bio株式会社)的缓冲液1mL中混合显色液100μL和消化液450μL,在激发356nm下测定458nm下的荧光强度(Infinite M200,Tecan Group Ltd.)。
将试剂盒所带的标准液(100μg/mL)进行2倍稀释制备DNA浓度的标准曲线。由该标准曲线的测定结果制成线性回归方程(Excel,Microsoft),算出样品的DNA浓度。
各个孔的放射活性用DNA含量进行标准化(cpm/μg DNA)。
其结果显示阳性对照的TGF-β1显示溶剂对照的10倍放射活性,蛋白多糖的合成量增加。PTH(1-34)显示溶剂对照的6倍放射活性。化合物7在10-6mol/L时显示溶剂对照的3倍放射活性,在10-5mol/L时显示溶剂对照的10倍放射活性(图5)。由该结果明显可知,化合物7在人关节软骨细胞中具有促进软骨基质的合成的作用。
实施例5:化合物7对兔半月板部分切除模型的效果
将12周龄的NZW系雄性兔(Oriental酵母工业株式会社)饲养驯化5天后,在异氟醚麻醉下切开左膝关节的外侧皮肤,切除外侧副韧带和芝麻韧带露出外侧半月板。将外侧半月板的中央部切除3-4mm的宽度,制成变形性关节病模型(Kikuchi T et al,Osteoarth Cart 1999;4(2):99-110.)。此时,将连接了在左侧大腿部皮下埋设的渗透泵3个(2ML1,Durect,Road Cupertino,CA,US)的聚乙烯管3根(PE60,日本Becton Dickinson株式会社)的前端留置在关节内,在关节内持续给予药液。在渗透泵中填充1)溶剂对照(50%二甲基亚砜/50%生理盐水,v/v)、2)化合物7 3.0μg/mL,3)化合物7 30μg/mL中的任一者。术后7天再次进行异氟醚麻醉,填充了与最初移植的相同的药剂的渗透泵进行交换。半月板部分切除术后14天将兔安乐死,采取大腿骨和下腿骨,浸渍并固定于20%中性缓冲福尔马林中。然后,将关节软骨表面的粗糙化用印度油墨进行染色(图7)。使用数码显微镜(VHX-2000,株式会社KEYENCE)对下腿骨的表面结构拍照,计测印度油墨阳性的病变部面积和外侧踝全体的面积,算出病变部的外侧踝全体所占的比例(图6)。其结果表面化合物7在用量依存性上病变部分面积变小。此时的下腿骨的关节软骨面的照片如图4所示。
实施例6:对正常大鼠反复经口给药4周下的对生长板软骨的效果
将从株式会社日本医科学动物资材研究所购入的雌性RccHan:WIST大鼠在、20~26℃、湿度30~70%的标准实验室条件下驯化1周以上后,用于实验。对于大鼠,自由摄取自来水以及含1.1%钙、1.0%磷酸和250IU/100g的维生素D3的标准啮齿动物饲料(CE-2、日本CLEA株式会社)。
测定6周龄的大鼠的体重,1组10只将大鼠分配进行分组,以使各组的平均体重达到均等。分组的第二天起对所有大鼠1天1次反复给药4周。媒介-对照组经口给予溶剂(媒介)。化合物7―6mg/kg、化合物7―60mg/kg、化合物7―600mg/kg组分别以6mg/kg、60mg/kg、600mg/kg的用量经口给予悬浮于媒介的化合物7。每组均以给药容量为2mL/kg进行给药。媒介使用丙二醇(特级、关东化学株式会社)。最终给药的第二天在麻醉下通过腹大动脉采取血液并将大鼠安乐死处置后,进行剖检,采取大腿骨。大腿骨用10%中性缓冲福尔马林液固定,除盐后制作石蜡包埋薄切组织标本(苏木-伊红染色)。将制作的标本的大腿骨远端用光学显微鏡进行病理组织学观察。结果示于表1,代表例的组织学的画像示于图8。
[表1]
对正常大鼠反复经口给药4周后的大腿骨生长板软骨的组织学的变化
如表1所示,化合物7组相对于媒介-对照组,在用量依存性上使大腿骨的生长板软骨肥厚。要求来自图8的组织画像以缩小比例尺为基础的代表例的生长板软骨的宽度(箭头标记)时,媒介-对照的个体约为390μm,化合物7―600mg/kg的个体约为2940μm。
如上所述化合物7的反复经口给予使大鼠的大腿骨的生长板软骨肥厚。这些作用基于化合物7的软骨同化作用的诱导、软骨的最终分化的抑制、或软骨的增殖亢进,可以认为化合物7的经口给予对变形性关节病的治疗有效。进而,对于通式(1)所示的化合物,在参考试验例1至5中可确认强的PTH样作用和高代谢稳定性,可期待通过基于PTH样作用的软骨同化作用对变形性关节病的治疗有效。
实施例7:对于向成熟龄的摘除卵巢大鼠反复给药3个月的骨密度的效果
将通过日本Charles River株式会社购入的雌性Crl:CD(SD)大鼠在20~26℃、湿度35~75%的标准试验室条件下驯化1周以上后,用于实验。对于大鼠,自由摄取自来水以及包含1.1%钙、1.0%磷酸和250IU/100g的维生素D3的标准啮齿动物饲料(CE-2)(日本CLEA株式会社)。
对8月龄的大鼠实施两侧的卵巣摘除术(OVX)及假手术(Sham)。在手术后第3个月测定体重和腰椎骨密度,实施分组,Sham组10只、OVX大鼠1组12只5组。分组后按照下述对各大鼠1天1次反复给药3个月。Sham-对照组和OVX对照组的大鼠分别通过经口给予(PO)需经口给予的溶剂(PO媒介)、通过静脉给予(IV)需静脉给予的溶剂(IV媒介)、通过皮下给予(SC)需皮下给予的溶剂(PC buffer)。向OVX-化合物7-PO组的的大鼠以30mg/kg的用量经口给予溶于PO媒介的上述化合物7,此外,IV媒介和PC buffer分别通过静脉给予和皮下给予。向OVX-化合物7-IV(低用量)组的大鼠以3mg/kg的用量静脉给予溶于IV媒介的上述化合物7,此外,PO媒介和PC buffer分别通过经口给予和皮下给予。向OVX-化合物7-IV(高用量)组的大鼠以10mg/kg的用量静脉给予溶于IV媒介的上述化合物7,此外,PO媒介和PC buffer分别通过经口给予和皮下给予。向OVX-hPTH(1-34)组的大鼠分别经口给予PO媒介、静脉给予IV媒介、以0.9nmol/kg的用量皮下给予溶于PC buffer的hPTH(1-34)。
此外,以与对人给予作为骨质疏松症治疗药在临床应用的Forteo(注册商标)20μg时相同程度的AUC(血中浓度―时间曲线下面积)的方式,将对大鼠给药时所显示的给药用量设定为0.9nmol/kg。每组均分别经口给予5mL/kg的用量、静脉给予2mL/kg的用量、皮下给予1mL/kg的用量。PO媒介或IV媒介使用将10%二甲基亚砜(和光纯药工业株式会社)、10%KolliphorEL(Sigma-Aldrich Japan合同会社)、10%羟基丙基‐β‐环糊精(日本食品化工株式会社)利用甘氨酸(和光纯药工业株式会社)及氢氧化钠(和光纯药工业株式会社)制备为pH10而得的组成。PC buffer使用将25mmol/L磷酸-枸橼酸缓冲液、100mmol/L NaCl、0.05%Tween80制备为pH5.0而得的组成。在最终给药的第二天在麻醉下通过腹大动脉采取血液而将大鼠安乐死处置后,进行剖检采取腰椎及大腿骨。
给药期间或者剖检时确认到与通常状态不同的大鼠从解析中除去。最终各组的只数为Sham-对照组9只、OVX-对照组12只、OVX-化合物7-PO组11只、OVX-化合物7-IV(低用量)组11只、OVX-化合物7-IV(高用量)组10只、OVX-hPTH(1-34)组12只。
腰椎和大腿骨保存于70%乙醇中。腰椎和大腿骨的骨密度使用双重X射线骨盐量测定装置(DCS-600EX、Aloka株式会社)测定。腰椎骨密度测定第2至第4腰椎,大腿骨骨密度测定是将大腿骨纵向10等分、测定膝侧远端的3份。将结果示于图10。
数据以平均值+标准偏差(SE)显示。使用JMP 9.02(SAS Institute Inc),实施以下的统计学解析。显著性水平为两侧5%。关于腰椎及大腿骨骨密度,通过2组的t检测进行Sham-对照组和OVX-对照组的比较(#P<0.05)。此外,将OVX-对照组作为对照组,进行相对于给予化合物7的3组以及给予hPTH(1-34)的1组共计4组的参数Dunnett型多重比较(*P<0.05)。
如图10所示,在大腿骨骨密度中,OVX-对照组相对于Sham-对照组显示出显著的骨密度减少,作为阳性对照的OVX-hPTH(1-34)组相对于OVX-对照组显示出显著的增加。OVX-hPTH(1-34)组相对于OVX-对照组的增加率为14.3%。此时OVX-化合物7-PO组和OVX-化合物7-IV(高用量)组相对于OVX-对照组显示出显著的增加,增加率分别为8.8%和18.7%。
此外,在腰椎骨密度中,OVX-对照组相对于Sham-对照组显示出显著的骨密度减少,作为阳性对照的OVX-hPTH(1-34)组相对于OVX-对照组显示出显著的增加。OVX-hPTH(1-34)组相对于OVX-对照组的增加率为22.1%。此时OVX-化合物7-IV(高用量)组相对于OVX-对照组显示出显著的增加,增加率为13.3%。
如上所述化合物7的反复经口给予或静脉给予时,骨质疏松症的病态模型即OVX大鼠的骨密度增加,因此认为化合物7对骨质疏松症、牙周病中的骨量减少、抜牙后的牙槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨再建的疾病的预防、治疗、改善、恢复促进有效。进而,对于通式(1)所示的化合物,在参考实施例2和5中可确认强的PTH样作用,认为通过基于PTH样作用的骨同化作用可得到骨密度增加作用。认为关于通式(1)所示的化合物,对骨质疏松症、牙周病中的骨量减少、抜牙后的牙槽骨损伤等、对需要骨同化作用的诱导、骨量的增加或骨再建的疾病的预防、治疗、改善、恢复促进有效。
实施例8:化合物7对兔半月板部分切除模型的效果
购入13周龄的Kb1:JW系雄性兔(Oriental酵母工业株式会社)饲养驯化,经过10天或11天后,在异氟醚麻醉下切开左膝关节的外侧皮肤,切除外侧副韧带和芝麻韧带露出外侧半月板。将外侧半月板的中央部切除3-4mm的宽度,制成变形性关节病模型(Kikuchi T et al,Osteoarth Cart1999)。半月板部分切除手术3天后,向膝关节内给予媒介或者化合物7的低用量(0.4mg)、中用量(2mg)、高用量(10mg)。媒介使用生理盐水,各个体均给予40μL。给予后28天后将兔子安乐死,采取大腿骨和胫骨,浸渍并固定于含有0.5%CPC的20%中性缓冲福尔马林中。然后,用10%EDTA液(pH7.4)除盐后,就大腿骨和胫骨的两侧踝的一定部位(膝窝筋起始部起5mm远端的大腿骨踝部横截面以及胫骨踝部中央部横截面)制作石蜡切片,并进行Safranin O染色(Safranin O、Fast Green和铁苏木素染色)。
使用光学显微镜(BHS、奥林巴斯光学工业株式会社)进行病理组织学检查。使用大腿骨外侧踝和胫骨外侧踝的一定部位的标本,对软骨病变进行定量评价,因此按照对Colombo等的评价法(Colombo C et.al,Arthritis Rheum.1983;26:875-86)进行了部分改变的Kikuchi等的评价标准(Kikuchi T et.al,Osteoarthritis and Cartilage.1995;4:99-110)、以+1~+4的4个阶段对表层的消失、软骨糜烂、粗糙化/皲裂、蛋白多糖染色性(Safranin O染色性)降低、软骨细胞序列不整、软骨细胞消失、软骨下骨露出、(软骨细胞的)房状集束形成这8个项目进行了评价。将8个项目的评价分数的总和作为综合组织学评分(简称为综合评分)(图11)。数据以平均值+标准偏差(SE)显示。病理组织学检查中的评价项目的各评分以及综合评分,使用统计软件SPSS 14.0J(日本IBM(株))进行Mann-Whitney的U检验。显著水平为危险率5以及1%。
其结果,在病理组织学检查时,胫骨外侧踝软骨病变中,与溶剂对照组相比,低用量组中的作为软骨病变综合指标的综合评分显示显著的低值(软骨变性抑制)。在中用量、高用量组中,也显示出综合评分的低值倾向。
由此认为,在胫骨外侧踝软骨病变中,化合物7具有软骨变性抑制作用。
参考实施例
用以下的参考实施例及参考试验例进一步详细地说明本发明的内容,但本发明不限于这些内容。所有的起始物质及试剂均为从商业供应商处获得,或使用公知的方法合成而得到。对于1H-NMR谱而言,使用Me4Si作为内标物质,或不使用内标物质,使用Mercury300(varian制)、ECP-400(JEOL制)、或400-MR(varian制)进行测定(s=单峰、d=双重峰、t=三重峰、brs=宽单峰、m=多重峰)。质谱分析使用质谱仪、ZQ2000(Waters制)、SQD(Waters制)、2020(Shimazu制)进行测定。
参考实施例1
1-(4-(2-((2-(4-氟-3-(三氟甲氧基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物1)。
(反应1-1)
在室温下,向4-溴-3,5-二甲基苯胺(3.47g,17.4mmol)和二异丙基乙基胺(5.3ml,30.4mmol)的DMI(13ml)溶液中,添加2-溴异丁酸(3.86g,23.1mmol)。在100℃下对混合物进行1小时加热搅拌。进而,添加2-溴异丁酸(496mg,2.97mmol)和二异丙基乙基胺(0.8ml,4.59mmol),然后100℃下对混合物进行1小时加热搅拌。
在室温下,向反应混合物中添加MeOH(52ml)和5N氢氧化钠水溶液(52ml,260mmol),然后在75℃下对该混合物进行1.5小时加热搅拌。将反应混合物冷却后,添加水,用1N硫酸氢钾水溶液调节至pH为5,用乙酸乙酯萃取。用水洗涤有机层后,用无水硫酸镁干燥,进行浓缩,作为粗产物得到2-((4-溴-3,5-二甲基苯基)氨基)-2-甲基丙酸(5.79g)。MS(ESI)m/z=286,288(M+H)+
(反应1-2)
在室温下,向2-((4-溴-3,5-二甲基苯基)氨基)-2-甲基丙酸(5.79g,反应1-1中得到的化合物)的二氯甲烷(62ml)和乙酸(62ml)混合物中,添加氰酸钠(5.03g,59.8mmol)。在室温下对混合物进行3小时搅拌。将反应混合物添加至饱和碳酸氢钠水溶液(400ml)中后,进一步用5N氢氧化钠水溶液调节至pH为7-8,用乙酸乙酯萃取。用无水硫酸镁干燥有机层,然后在减压下浓缩。用乙酸乙酯-己烷、二氯甲烷-己烷依次洗涤得到的固体,得到了1-(4-溴-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(3.80g,66%)。
MS(ESI)m/z=311,313(M+H)+
(反应1-3)
在氮气流下,在110℃下,对8-(乙烯基磺酰基)-1,4-二氧杂-8-氮杂螺[4.5]癸烷(431mg,1.85mmol)、1-(4-溴-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(575mg,1.85mmol)、三(二苄叉丙酮)二钯(0)(508mg,0.55mmol)、四氟硼酸三叔丁基膦(165mg,0.55mmol)和甲基二环己基胺(2.1ml,9.25mmol)的N-甲基-2-吡咯烷酮(18.5ml)混合物进行2小时搅拌。将反应混合物冷却后,用水猝灭,用乙酸乙酯萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸镁干燥,在减压下浓缩。用氨基硅胶柱色谱法(二氯甲烷-甲醇)纯化得到的残渣,得到(E)-1-(4-(2-(1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-基磺酰基)乙烯基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(584mg,68%)。
MS(ESI)m/z=464(M+H)+
(反应1-4)
向(E)-1-(4-(2-(1,4-二氧杂-8-氮杂螺[4.5]癸烷-8-基磺酰基)乙烯基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(1.2g,2.58mmol)的四氢呋喃(26ml)溶液中,经10分钟滴加2N盐酸水溶液(26ml,52mmol)。在60℃下对混合物进行2小时加热搅拌。将反应混合物冷却后,用2N氢氧化钠水溶液调节至pH为7,用乙酸乙酯萃取。将有机层用饱和食盐水洗涤后,用无水硫酸镁干燥,在减压下浓缩。用硅胶柱色谱法(二氯甲烷-乙酸乙酯)纯化得到的残渣,得到(E)-1-(3,5-二甲基-4-(2-((4-氧代哌啶-1-基)磺酰基)乙烯基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(998mg,92%)。
MS(ESI)m/z=420(M+H)+
(反应1-5)
在室温下,向(E)-1-(3,5-二甲基-4-(2-((4-氧代哌啶-1-基)磺酰基)乙烯基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(994mg,2.37mmol)的甲醇(24ml)溶液中,依次添加氰化钾(188mg,2.84mmol)和乙酸铵(237mg,3.08mmol)。在60~70℃下对混合物进行3小时加热搅拌。将反应混合物冷却后,在减压下浓缩,用乙酸乙酯稀释。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸镁干燥,在减压下浓缩。用硅胶柱色谱法(二氯甲烷-乙酸乙酯)纯化得到的残渣,得到(E)-4-氨基-1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙烯基)磺酰基)哌啶-4-甲腈(681mg,68%)。
1H-NMR(300MHz,DMSO-d6)δ:1.3(6H,s),1.7(2H,m),2.0(2H,m),2.3(6H,s),2.7(2H,s),2.9(2H,m),3.4(2H,m),6.9(1H,d,J=15.9Hz),7,1(2H,s),7.4(1H,d,J=15.9Hz),11.2(1H,brs)
(反应1-6)
在室温下,向(E)-4-氨基ー1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙烯基)磺酰基)哌啶-4-甲腈(675mg,1.50mmol)的甲醇(7.5ml)和二甲基亚砜(0.195ml)溶液中,依次缓慢滴加2N氢氧化钠水溶液(1.6ml,1.6mmol)和30%过氧化氢水溶液(0.2ml,1.95mmol)。在室温下对混合物进行1小时搅拌。向反应混合物中添加乙酸乙酯、己烷、饱和氯化铵水溶液。滤取析出的固体,进行洗涤、干燥,得到(E)-4-氨基-1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙烯基)磺酰基)哌啶-4-甲酰胺(498mg,72%)。
MS(ESI)m/z=464(M+H)+
(反应1-7)
在氢气氛下,在室温下,对(E)-4-氨基-1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙烯基)磺酰基)哌啶-4-甲酰胺(1.3g,2.8mmol)和氢氧化钯/碳(Pd20%)(约50%水湿润品)(1.3g)的甲醇(21ml)-二甲基甲酰胺(7ml)混合物进行4小时搅拌。将反应混合物过滤、洗涤,然后在减压下浓缩滤液,得到4-氨基-1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙基)磺酰基)哌啶-4-甲酰胺(998mg,77%)。
MS(ESI)m/z=466(M+H)+
(反应1-8)
向4-氨基-1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙基)磺酰基)哌啶-4-甲酰胺(120mg,0.258mmol)、4-氟-3-(三氟甲氧基)苯甲酸(69mg,0.309mmol)和二异丙基乙基胺(0.09ml,0.516mmol)的二甲基甲酰胺(2.5ml)溶液中,添加O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐(HATU)(118mg,0.309mmol)。在室温下对混合物进行1.5小时搅拌。用水将反应混合物猝灭,然后用二氯甲烷进行萃取。将有机层用饱和食盐水洗涤后,用无水硫酸钠洗涤后,在减压下浓缩,得到1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙基)磺酰基)-4-(4-氟-3-(三氟甲氧基)苯甲酰胺)哌啶-4-甲酰胺(150mg,67%)。
MS(ESI)m/z=672(M+H)+
(反应1-9)
在0℃下,向1-((4-(5,5-二甲基-2,4-二氧代咪唑烷-1-基)-2,6-二甲基苯乙基)磺酰基)-4-(4-氟-3-(三氟甲氧基)苯甲酰胺)哌啶-4-甲酰胺(150mg,0.223mmol)的叔丁醇(2.5ml)和乙醇(2.5ml)的混合液中,添加叔丁醇钾(75mg,0.670mmol)。在氮气流下、在50℃下对混合物进行1.5小时加热搅拌。将反应混合物冷却后,用水稀释,用饱和氯化铵水溶液猝灭,用二氯甲烷萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩。用硅胶柱色谱法(二氯甲烷-甲醇)纯化得到的残渣,得到1-(4-(2-((2-(4-氟-3-(三氟甲氧基)苯基)-4-氧代-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(118mg,81%)。
MS(ESI)m/z=654(M+H)+。1H-NMR(400MHz,CD3OD)δ:1.40(6H,s),1.71-1.80(2H,m),2.00-2.08(2H,m),2.43(6H,s),3.22(4H,s),3.47-3.57(2H,m),3.80-3.88(2H,m),7.01(2H,s),7.50-7.57(1H,m),7.97-8.04(1H,m),8.05-8.12(1H,m)
使用适当的羧酸起始原料、试剂、溶剂,通过与参考实施例1的反应1-8、反应1-9同样的操作,合成以下所示的参考实施例化合物。
(化合物2-5)
[表2]
参考实施例2
1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物6)。
(反应2-1)
在氮气流下,在100℃下,对按照WO2010/126030(A1)的路线2、路线3及路线12中记载的方法合成的2-(3-(三氟甲基)苯基)-8-(乙烯基磺酰基)-1,3,8-三氮杂螺[4.5]癸-1-烯-4-酮(150mg,0.387mmol)、1-(4-溴-3,5-二甲基苯基)-5,5-二甲基咪唑烷-2,4-二酮(169mg,0.542mmol)、双(二苄叉丙酮)钯(45mg,0.077mmol)、四氟硼酸三叔丁基膦(22mg,0.077mmol)和甲基二环己基胺(0.123ml,0.581mmol)的N-甲基-2-吡咯烷酮(0.97ml)混合物进行1小时加热搅拌。将反应混合物冷却后,用水猝灭,用乙酸乙酯萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩。用硅胶柱色谱法(乙酸乙酯-己烷)纯化得到的残渣,得到了(E)-1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙烯基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(197mg,82%)。
MS(ESI)m/z=618(M+H)+。
(反应2-2)
在氢气氛下,在室温下,对(E)-1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙烯基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(195mg,0.316mmol)和氢氧化钯/碳(Pd20%)(约50%水湿润品)(195mg,0.139mmol)的2,2,2-三氟乙醇(6ml)混合物进行14小时搅拌。将混合物过滤后,在减压下浓缩滤液。用硅胶柱色谱法(乙酸乙酯-己烷)纯化得到的残渣,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(121mg,62%)。
MS(ESI)m/z=620(M+H)+。1H-NMR(400MHz,CD3OD)δ:1.40(6H,s),1.72-1.81(2H,m),2.00-2.10(2H,m),2.44(6H,s),3.22(4H,s),3.50-3.58(2H,m),3.80-3.88(2H,m),7.01(2H,s),7.72-7.79(1H,m),7.88-7.94(1H,m),8.16-8.23(1H,m),8.31(1H,s)
参考实施例3
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物7)。
(反应3)
使用适当的起始原料、溶剂,利用与参考实施例2同样的操作,合成1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物7)。
MS(ESI)m/z=636(M+H)+。1H-NMR(400MHz,CDCl3)δ:1.47(6H,s),1.70-1.78(2H,m),2.10-2.19(2H,m),2.40(6H,s),3.00-3.07(2H,m),3.19-3.25(2H,m),3.45-3.53(2H,m),3.81-3.88(2H,m),6.94(2H,s),7.35(2H,d,J=8.0Hz),7.73(1H,brs),7.93(2H,d,J=8.0Hz),9.37(1H,brs)
参考实施例4
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮(化合物8)。
(反应4-1)
在室温下,向环戊酮(42mg,0.500mmol)和4-溴-3,5-二甲基苯胺(100mg,0.500mmol)的乙酸(0.5ml)混合物中,添加三甲基氰硅烷(0.063ml,0.500mmol)。在氮气流下,在室温下,对混合物进行1.5小时搅拌。将反应混合物放入到28%氨水(1ml)中将其猝灭后,用水稀释,用二氯甲烷萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩,作为粗产物得到1-((4-溴-3,5-二甲基苯基)氨基)环戊烷甲腈(152mg)。
1H-NMR(400MHz,CDCl3)δ:1.83-1.92(4H,m),2.07-2.15(2H,m),2.33-2.42(2H,m),2.37(6H,m),3.71(1H,brs),6.56(2H,s)
(反应4-2)
在室温下,向1-((4-溴-3,5-二甲基苯基)氨基)环戊烷甲腈(145mg,0.495mmol)的二氯甲烷(5ml)溶液中,添加2,2,2-三氯乙酰基异氰酸酯(0.070ml,0.593mmol)。在氮气流下,在室温下对混合物进行1小时搅拌。
向反应混合液中,依次添加三乙基胺(0.103ml,0.742mmol)、水(0.045ml)及甲醇(0.10ml)后,在氮气流下对混合物进行1.5小时加热回流。将反应混合物冷却后,用水稀释,用1N盐酸水溶液调节至pH为5后,用二氯甲烷萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩,作为粗产物得到1-(4-溴-3,5-二甲基苯基)-4-亚氨基-1,3-二氮杂螺[4.4]壬烷-2-酮。
MS(ESI)m/z=336,338(M+H)+。
(反应4-3)
在氮气流下,在65℃下对1-(4-溴-3,5-二甲基苯基)-4-亚氨基-1,3-二氮杂螺[4.4]壬烷-2-酮(前反应中得到的粗产物)的乙酸(1.0ml)和水(0.25ml)的混合物进行1.5小时加热搅拌。进而,添加乙酸(1.0ml)和水(0.25ml)后,在氮气流下,在65℃下对混合物进行17小时加热搅拌。将反应混合物冷却后,用水稀释,用饱和碳酸氢钠水溶液调节至pH为8,用乙酸乙酯萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩。用硅胶柱色谱法(乙酸乙酯-己烷)纯化得到的残渣,得到1-(4-溴-3,5-二甲基苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮(121mg)。
MS(ESI)m/z=337,339(M+H)+。
(反应4-4)
使用适当的起始原料、溶剂,利用与参考实施例2同样的操作,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮(化合物8)。
MS(ESI)m/z=662(M+H)+。1H-NMR(400MHz,DMSO-d6)δ:1.36-1.44(2H,m),1.60-1.70(4H.m),1.82-1.91(2H,m),1.91-2,06(4H,m),2.38(6H,s),3.01-3.09(2H,m),3.22-3.30(2H,m),3.30-3.42(2H,m),3.70-3.77(2H,m),7.03(2H,s),7.57(2H,d,J=8.4Hz),8.14(2H,d,J=8.4Hz)
参考实施例5
1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-8-甲基-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮(化合物9)。
(反应5-1)
使用4-氧代哌啶-1-甲酸叔丁酯作为起始原料,另外,使用适当的溶剂,利用与参考实施例4同样的操作,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2,4-二氧代-1,3,8-三氮杂螺[4.5]癸烷-8-甲酸叔丁酯。
MS(ESI)m/z=777(M+H)+。
(反应5-2)
在室温下,向1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2,4-二氧代-1,3,8-三氮杂螺[4.5]癸烷-8-甲酸叔丁酯(11.7mg,0.015mmol)的二氯甲烷(0.13ml)混合液中,添加三氟乙酸(0.05ml,0.673mmol)。在氮气流下,在室温下对混合物进行1小时搅拌。在减压下浓缩反应混合物,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮2三氟乙酸盐(13.6mg)。
MS(ESI)m/z=677(M+H)+。
(反应5-3)
向1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮2三氟乙酸盐(21.1mg,0.022mmol)的甲酸(0.033ml)混合物中,添加37%甲醛水溶液(0.055ml)。在氮气流下,在80℃下对混合物进行3小时加热搅拌。将反应混合物浓缩后,用乙酸乙酯稀释残渣。用稀氢氧化钠水溶液洗涤有机层后,用无水硫酸镁干燥,在减压下浓缩。用柱色谱法(二氯甲烷-甲醇)纯化得到的残渣,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-8-甲基-1,3,8-三氮杂螺[4.5]癸烷-2,4-二酮(4.5mg,30%)。
MS(ESI)m/z=691(M+H)+。1H-NMR(400MHz,CD3OD)δ:1.76-1.84(2H,m),1.92-2.02(2H,m),2.02-2.12(4H,m),2.38(3H,s),2.46(6H,s),2.81-2.88(2H,m),2.92-3.02(2H,m),3.23(4H,s),3.51-3.60(2H,m),3.72-3.80(2H,m),7.01(2H,s),7.48(2H,d,J=8.0Hz),8.10(2H,d,J=8.0Hz)
参考实施例6
5-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2-氧杂-5,7-二氮杂螺[3.4]辛烷-6,8-二酮(化合物10)。
(反应6)
使用氧杂环丁烷-3-酮作为起始原料,另外,使用适当的溶剂,利用与参考实施例4同样的操作,得到5-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-2-氧杂-5,7-二氮杂螺[3.4]辛烷-6,8-二酮。
MS(ESI)m/z=650(M+H)+。1H-NMR(400MHz,CDC13)δ:1.69-1.77(2H,m),2.12-2.22(2H,m),2.45(6H,s),3.03-3.11(2H,m),3.22-3.29(2H,m),3.46-3.53(2H,m),3.84-3.91(2H,m),4.86(2H,d,J=7.2Hz),5.03(2H,d,J=7.2Hz),7.07(2H,s),7.35(2H,d,J=8.4Hz),7.98(2H,d,J=8.4Hz),8.56(1H,s),10.34(1H,s)
参考实施例7
4-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-4,6-二氮杂螺[2.4]庚烷-5,7-二酮(化合物11)。
(反应7-1)
在氮气流下,在120℃下,对2-溴-5-碘-1,3-二甲基苯(300mg,0.965mmol)、1-氨基环丙烷甲酸(195mg,1.93mmol)、碘化铜(I)(37mg,0.194mmol)和二氮杂双环十一碳烯(0.50ml,3.35mmol)的二甲基乙酰胺(2.6ml)混合物进行3小时加热搅拌。用硅胶柱色谱法(Wakosil C18,乙腈-水(0.1%甲酸)纯化反应混合物,得到1-((4-溴-3,5-二甲基苯基)氨基)环丙烷甲酸(219mg,80%)。
MS(ESI)m/z=284,286(M+H)+。
(反应7-2)
在室温下,向1-((4-溴-3,5-二甲基苯基)氨基)环丙烷甲酸(198mg,0.697mmol)的乙酸(3ml)和二氯甲烷(1.5ml)混合物中,添加氰酸钾(424mg,5.23mmol)。在室温下对混合物进行1小时搅拌后,在60℃下进行2小时加热搅拌。用饱和碳酸氢钠水溶液将反应混合物调节至pH为8后,用乙酸乙酯萃取。将有机层依次用水、饱和食盐水洗涤后,用无水硫酸钠干燥,在减压下浓缩。用硅胶柱色谱法(乙酸乙酯-己烷)纯化得到的残渣,得到4-(4-溴-3,5-二甲基苯基)-4,6-二氮杂螺[2.4]庚烷-5,7-二酮(49mg,23%)。
MS(ESI)m/z=309,311(M+H)+。
(反应7-3)
使用适当的起始原料、溶剂,利用与参考实施例2同样的操作,得到4-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-4,6-二氮杂螺[2.4]庚烷-5,7-二酮(化合物11)。
MS(ESI)m/z=634(M+H)+。1H-NMR(400MHz,DMSO-d6)δ:0.99-1.03(2H,m),1.19-1.27(4H,m),1.58-1.64(2H,m),1.81-1.90(2H,m),2.35(6H,s),2.99-3.04(2H,m),3.22-3.29(2H,m),3.67-3.73(2H,m),6.95(2H,s),7.56(2H,d,J=8.4Hz),8.12(2H,d,J=8.4Hz)
参考实施例8
1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮(化合物12)。
(反应8)
使用适当的起始原料、溶剂,利用与参考实施例2同样的操作,得到1-(3,5-二甲基-4-(2-((4-氧代-2-(3-(三氟甲基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-1,3-二氮杂螺[4.4]壬烷-2,4-二酮。
MS(ESI)m/z=646(M+H)+。1H-NMR(400MHz,DMSO-d6)δ:1.40-1.48(2H,m),1.62-1.71(4H,m),1.88-1.97(2H,m),1.97-2.08(4H,m),2.41(6H,s),3.03-3.10(2H,m),2.29-3.34(2H,m),3.38-3.47(2H,m),3.72-3.79(2H,m),7.06(2H,s),7.84(1H,dd,J=7.6,7.6Hz),8.02(1H,d,J=7.6Hz),8.33(1H,d,J=7.6Hz),8.38(1H,s)
[参考试验例]
将针对本发明的化合物的通过人PTH1R进行的cAMP产生能力试验、通过大鼠受体进行的cAMP产生能力试验、使用人肝微粒进行的代谢稳定性试验、使用大鼠肝细胞进行的代谢稳定性试验、使用TPTX大鼠模型进行的血清Ca浓度上升作用试验等各试验的结果示于参考试验例1~5。应予说明,作为比较化合物,使用表3所示的WO2010/126030A1中记载的化合物。
[表3]
参考试验例1:人PTH1R中的化合物的体外cAMP信号活性的测定(肽)人PTH(1-34)及降钙素从肽研究所(日本大阪)购入,将其溶解于10mM乙酸中,成为1mM,在-80℃冰箱中保存。
(细胞培养)
在添加有10%胎牛血清(Hyclone)、100单位/ml青霉素G、及100μg/ml硫酸链霉素(Invitrogen Corp)的达尔伯克氏改良伊格尔氏培养基(DMEM)中,在含有5%CO2的加湿气氛下,在37℃下培养细胞。
将未表达PTH1R的LLC-PK1细胞、及每1个LLC-PK1细胞过表达9.5×105个人PTH1R的HKRK-B7用于cAMP信号传递分析(Takasu et al.,J.Bone.Miner.Res.14:11-20,1999)。
(cAMP刺激)
以1×105细胞/孔将HKRK-B7或LLC-PK1细胞接种于96孔板,孵育一夜。第二天,然后添加含有人PTH(1-34)或化合物的50μl的cAMP分析缓冲液(DMEM、2mM IBMX、0.2mg/ml牛血清白蛋白、35mM Hepes-NaOH、pH7.4),放置在在37℃的培养箱中,孵育20分钟。除去培养基后,用100μl的cAMP分析缓冲液洗涤细胞1次。为了将细胞冷冻,将培养板放在干冰粉末上,然后从干冰中取出。使用40μl的50mM HCl将细胞熔化,再次在干冰上冷冻。使用市售的cAMP EIA试剂盒(Biotrack cAMP EIA system,GE health care),测定细胞内cAMP的产生量。
(体外cAMP诱导能力的测定中的20%有效浓度(EC20)及50%有效浓度(EC50)的计算。)
使用基于可变梯度的S型用量反应式进行分析,将100nM时的人PTH(1-34)的cAMP信号活性作为100%,将各化合物显示20%或50%的cAMP信号活性的浓度作为EC20或EC50来进行计算。
HKRK-B7细胞中的结果示于表4。
应予说明,LLC-PK1细胞中的cAMP应答的程度比HKRK-B7细胞中的程度低。
[表4]
参考试验例2:大鼠PTH1R中的化合物的体外cAMP信号活性的测定
代替HKRK-B7细胞,使用中外制药建立的过表达大鼠PTH1R的LLC-PK46_RATO_PTH1R细胞,与参考试验例1同样地进行测定。
将使用LLC-PK46_RATO_PTH1R细胞的结果示于表5。
大鼠PTH1受体中的体外cAMP信号活性的EC20值,与人PTH1R中的体外cAMP信号活性的EC20值之间存在良好的相关关系。关于EC50值,在大鼠与人之间存在良好的相关关系。
[表5]
参考试验例3:使用了人肝微粒的代谢稳定性试验
在0.1M磷酸缓冲液(pH7.4)中,在NADPH共存下,在37℃下孵育人肝微粒体和化合物或比较例规定的时间。使用LC/MS/MS测定各反应时间的母体化合物浓度,从残留率相对于反应时间的斜率中算出固有清除率(μL/min/mg protein)。
<分析条件>
化合物浓度:1μM
微粒体:0.5mg/mL
NADPH:1mM
反应时间:0、5、15及30分钟。
结果示于表6。化合物1~11与比较例1~6相比,相对于人肝微粒显示高代谢稳定性。
[表6]
参考试验例4:使用了大鼠肝细胞的代谢稳定性试验
利用胶原酶回流法由大鼠(SD、♀)的肝脏制备肝细胞。添加参考实施例化合物或比较例,在37℃下孵育规定的时间,然后添加反应终止液。使用LC/MS/MS测定各反应时间的母体化合物浓度,从残留率相对于反应时间斜率中算出固有清除率(μL/106cells/min)。
<分析条件>
细胞浓度:1×106cells/mL
化合物浓度:1μM
培养基:Williams'medium E
反应时间:0、15、30、60、120及240分钟
反应终止液:乙腈/2-丙醇(4/6、v/v)。
结果示于表7。化合物2、4、5、6、7、8、9、10、11的化合物与比较例1、2、3、5、6相比,大鼠肝细胞代谢稳定性提高。
[表7]
参考试验例5:TPTX大鼠模型中的血清Ca浓度上升作用
从日本Charles River株式会社(厚木饲育中心)获得4周龄的雌性Crl:CD(SD)大鼠,在20~26℃、湿度35~75%的标准实验室条件下,驯化1周。使大鼠自由摄取自来水以及含有1.1%钙、1.0%磷酸及250IU/100g的维生素D3的标准啮齿动物饲料(CE-2)(日本CLEA株式会社)。
对5周龄的大鼠施予TPTX。对一部分个体实施假手术(Sham)。对于用于使用的TPTX大鼠,选择手术4天后的血清Ca浓度小于8mg/dL的个体。手术5天后,基于手术4天后测得的血清Ca浓度和体重,分成8个TPTX组和1个Sham组,每组5只。以10mL/kg的用量向Sham组及TPTX-媒介组仅施予溶剂。向TPTX-各分析物组,分别以30mg/10mL/kg的用量将各分析物溶解到溶剂中并进行经口给予。对于溶剂的组成而言,使用以下溶剂:通过10%二甲基亚砜(和光纯药工业株式会社)、10%Cremophor EL(Sigma-Aldrich Japan合同会社)、20%羟丙基‐β‐环糊精(日本食品化工株式会社)、甘氨酸(和光纯药工业株式会社)制备成pH为10的溶剂。对于各组,在即将给予之前,进行预采血(Pre采血),在给予2、6、10及24小时后,也实施采血,测定血清Ca浓度。各组血液采集在异氟烷吸入麻醉下从颈静脉进行。
血清的Ca的测定。使用自动分析装置TBA-120FR(TOSHIBA MEDICAL SYSTEMS株式会社)测定通过离心分离从采集的血液获得的血清。
对动物试验进行统计分析。将数据表示为平均值±标准误差(SE)。统计的显著性使用SAS前临床包(Ver.5.00.010720、SAS Institute Japan,Tokyo,Japan)来确定。将小于0.05的p值视为统计学显著。通过2组的t检验显示:相对于TPTX‐媒介组,#P<0.05,相对于比较例1的组,*P<0.05,相对于比较例2的组,∫P<0.05。
对于血清Ca浓度的Pre值而言,Sham组为9.9mg/dL,TPTX各组为5.3~6.2mg/dL。关于给予各化合物后直到24小时为止的血清Ca浓度,将相对于Pre值的变化量的平均值示于图9。另外,对于给予各化合物后的血清Ca浓度的峰而言,在所有化合物中均为给予6小时后或10小时后。
对于大鼠肝细胞代谢稳定性高的化合物6、化合物7、化合物8而言,相对于pre值的正的变化量大,在经口给予时确认了强血清Ca浓度上升作用。另一方面,大鼠肝细胞代谢稳定性低的化合物1、比较例1、比较例2的相对于Pre值的正的变化量比化合物6、化合物7、化合物8小。尤其是,化合物7、化合物8相对于比较例1、比较例2统计显著。
此外,对于大鼠肝细胞代谢稳定性高的化合物6、化合物7、化合物8而言,在给予后6小时或10小时后,作为各化合物的最大值,显示7.8~8.5mg/dL的值,达到甲状旁腺功能减退患者的血清Ca浓度的治疗目标范围7.6~8.8mg/dL。另一方面,大鼠肝细胞代谢稳定性低的化合物1、比较例1、比较例2在所有的测定时间内,均未达到该治疗目标范围。
由以上的试验结果确认,在向大鼠经口给予化合物6、化合物7、化合物8(在强制表达了大鼠PTH1R的细胞中,显示强cAMP信号活性,并且相对于大鼠肝细胞中的代谢,显示高稳定性)时,确认了强血清Ca浓度上升作用。这些化合物在强制表达人PTH1R的细胞中具有cAMP信号活性,人肝微粒代谢稳定性比比较化合物高,在经口给予时,可期待针对甲状旁腺功能减退患者的良好治疗效果。进而,关于与化合物6、化合物7、化合物8同等程度地、在强制表达人PTH1R的细胞中显示cAMP信号活性并且显示人肝微粒代谢稳定性的通式(1)所示的化合物,也可期待针对甲状旁腺功能减退患者的良好治疗效果。
工业适用性
通过本发明,可提供通过将具有高代谢稳定性且发挥强PTH样作用的乙内酰脲衍生物非侵入地全身暴露或局部暴露而诱导骨/软骨同化作用,用于促进骨质疏松症、牙周病中的骨量减少、拔牙后的牙槽骨损伤、变形性关节病、关节软骨损伤、无动力性骨病、软骨成长不全症、低软骨形成症、骨质软化病、骨折等的预防、治疗、恢复和治愈的药物。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- 一种新型罗汉果醇衍生物单体的制作方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法