一种大分子前药纳米药物、制备方法及其应用与流程
本发明是一种具有肿瘤治疗作用的大分子前药纳米药物及其制备方法和用途,涉及医药技术领域。
背景技术:
许多小分子化学药物因为疏水性太强或者亲水性太强,亲脂性太差,难以透过细胞膜,导致治疗疾病的效果差。
一些具有强烈生理活性的植物来源药物具有强烈疏水性,给药困难,应用受限。喜树碱(camptothecin,cpt)和10-羟基喜树碱(hcpt)具有较强细胞毒活性。喜树碱类化合物用于肿瘤治疗的最大缺陷是水溶性很差,难以获得高有效浓度的溶液。为了改善cpt、hcpt的疏水性缺陷,有研究机构和企业对喜树碱的化学结构进行改性,合成了数百种喜树碱衍生物。至今已经有2种具有一定亲水性的喜树碱衍生物被批准用于临床肿瘤治疗,分别是伊立替康和拓扑替康,但其水溶性仍然较低。
紫杉醇也是一种生物碱,是目前唯一一种可以促进微管聚合和稳定已聚合微管的药物。研究表明,紫杉醇结合到聚合的微管上,不与未聚合的微管蛋白二聚体反应。紫杉醇主要适用于卵巢癌和乳腺癌,对肺癌、大肠癌、黑色素瘤、头颈部癌、淋巴瘤、脑瘤也都有一定疗效。但是,紫杉醇水溶性差,需采用聚氧乙烯蓖麻油和乙醇助溶,而聚氧乙烯蓖麻油可能导致过敏反应,患者用药前需预服抗过敏药物。研究表明,聚氧乙烯蓖麻油的应用降低了紫杉醇的抗肿瘤活性。同时,紫杉醇由于选择性差,有较强的毒性作用,易产生耐药性,以及无法透过血脑屏障等缺陷。
将药物包裹到载体中进行控制释放是一种重要的给药方式。纳米药物是将药物分子包埋在聚合物纳米载体中,具有诸多优势:1)显著提高疏水性药物的溶解性,延长药物体内的循环时间,提高生物利用度;2)通过被动靶向(epr效应)或主动靶向使药物特异性地靶向肿瘤组织,降低其在正常组织中的分布,从而降低药物毒副作用。用于治疗多种癌症的抗癌药doxil是包埋阿霉素的脂质体药物,表面有聚乙二醇(peg)水化层。临床证明,doxil可以延长药物在体内的循环时间,并且使药物在肿瘤部位富集,减少药物的心脏毒性。此外,多个基于peg的高分子纳米药物已进入临床或不同临床试验阶段。例如,聚乙二醇-聚乳酸共聚物(peg-pla)负载紫杉醇(ptx)的纳米胶束药物(genexol-pm)自2007年已在韩国用于临床治疗乳腺癌、肺癌和卵巢癌等。
但是,现有纳米药物还面临众多挑战:1)纳米药物包载效率低(<10%),注射到体内后,由于被稀释及与血液中不同成分的作用而很容易分解或集聚,导致包载的药物泄漏或过早释放;2)peg纳米药物在进入肿瘤组织后药物释放速度太慢或释放不充分,药物利用度低。
因此,有必要开发新的纳米药物释放系统,提高药物溶解性、稳定性、靶向性,并赋予还原敏感快速释药的功能,从而达到降低毒副作用、提高生物利用度的目的。
技术实现要素:
技术问题:本发明提供一种大分子前药纳米药物和制备方法及其应用,疏水性药物分子通过双硫键与低分子量聚乙二醇亲水高分子相连形成大分子前药,前药组装成纳米药物,并通过双硫键交联形成内核,外壳是低分子量聚乙二醇亲水高分子刷,纳米药物具有还原敏感靶向肿瘤细胞的释药功能,以及更好的稳定性,不发生药物泄漏。
技术方案:本发明的大分子前药纳米药物,包括两个疏水性药物分子由双硫键通过硫辛酸与低分子量聚乙二醇相连形成的通式(1)的两亲性大分子前药,所述两亲性大分子前药经过自组装并通过分子间双硫键交联形成内核含有药物、外壳是低分子量聚乙二醇亲水高分子刷的纳米颗粒,尺寸为10~1000纳米:
式中,x是丝氨醇或3-氨基甘油,d是疏水性药物,y是ch2ch2ocoo或ch2ch2oconh,n为6~80。
本发明纳米药物的优选方案中,上述疏水性药物是紫杉醇、多西他赛、卡巴他赛、阿霉素、表阿霉素、柔红霉素、去甲氧基柔红霉素、阿柔比星、吡柔比星、佐柔比星、喜树碱、7-乙基-10-羟基喜树碱、羟基喜树碱、拓扑替康、伊立替康、鲁比替康、贝洛替康、三尖杉酯碱、异三尖杉酯碱、雷帕霉素、美登素、依维莫司或达沙替尼。
本发明纳米药物的优选方案中,纳米药物还包括药效学上可接受的载体。
本发明纳米药物的优选方案中,纳米药物还包括助剂。进一步的优选方案中,助剂是脂肪或磷脂。
本发明的纳米药物优选方案中,纳米药物是液体制剂、固体制剂、半固体制剂、胶囊剂、颗粒剂、凝胶剂、注射剂、缓释制剂或控释制剂。
从体外药理活性等筛选来看,本发明大分子前药纳米药物表现良好的抗肿瘤等生物活性。试验表明本发明大分子前药纳米药物的体内毒性小于原药。因此可作为抗肿瘤等药物用于病人。
本发明的大分子前药纳米药物的制备方法,是将通式(1)的大分子前药在水溶液中组装成直径为10-1000纳米的纳米胶束,加入过氧化氢或在空气中巯基氧化导致分子间交联,形成内核含有疏水药物,外层为亲水性聚乙二醇刷的交联纳米颗粒,经冷冻干燥得到包裹药物的交联纳米颗粒冻干粉。
本发明还提供一种上述大分子前药纳米药物在制备抗肿瘤药物中的应用。
本发明的大分子前药纳米药物的制备方法,是由本发明的大分子前药或本发明大分子前药与助剂的混合物,通过薄膜分散法、逆相蒸发法、冷冻干燥法、超声波分散法、喷雾干燥法、膜挤压法、或高压均质法等方法制备。
本发明利用大分子前的两亲性,自组装成纳米颗粒,利用大分子中的巯基交联形成内核,低分子量聚乙二醇刷构成外壳,制备成内核交联,外壳亲水的纳米药物,提高纳米药物的药物负载率、稳定性;本发明的纳米药物,半衰期长,抗肿瘤等活性显著优于原药。
本发明的大分子前药纳米药物不仅是一种前药,也是药物载体,具有长循环、靶向肿瘤、敏感释放功能,具有低毒副作用。
有益效果:本发明与现有技术相比,具有以下优点:
本发明的大分子前药纳米药物,其前药分子中仅有两个药物分子基团,亲水部分为低分子量的聚乙二醇,前药分子分子量低,结构清晰。
本发明的大分子前药纳米药物,利用大分子前药的两亲性,自组装成纳米颗粒,利用大分子前药分子中的巯基交联形成内核,低分子量聚乙二醇刷构成外壳,制备成内核交联,外壳亲水的纳米药物,药物负载率高。
本发明的大分子前药纳米药物,外壳为低分子量聚乙二醇刷,纳米颗粒稳定性好,体内循环半衰期长。
本发明的大分子前药纳米药物,疏水性药物分子通过双硫键键合到高分子链上,药物分子不会泄漏,具有谷胱甘肽还原敏感性,主要在肿瘤组织或肿瘤细胞内快速释药,正常组织中药物分子难以释放,具有肿瘤靶向性。
本发明的大分子前药纳米药物,大分子前药通过双硫键交联形成内核,在体内不易解体,同时具有还原敏感性,在肿瘤组织或肿瘤细胞内快速解体,实现快速释药。
本发明的大分子前药纳米药物,大分子前药采用低分子量聚乙二醇,在体内降解后释放出的低分子量的聚乙二醇在体内停留时间短,易于快速排出体外,不在体内滞留,体内不产生聚乙二醇抗体。
本发明的大分子前药纳米颗粒,克服了物理包裹型纳米药物中药物易于泄漏的缺点。
本发明的大分子前药纳米药物的含有药物的部分包含双硫键,可以被谷胱甘肽等降解而快速断裂,断裂后含有药物分子的碎片中的巯基等通过“反咬”进攻分子自身的羰基,导致快速释放原药,克服了普通间隔臂难以快速断裂而不能快速释放原药的缺陷,具有更强的药效。
本发明的大分子前药纳米药物具有被动靶向作用,实现靶向释放。
本发明的大分子前药纳米药物,可用作液体制剂、固体制剂、半固体制剂。
本发明的大分子前药纳米药物结合靶向基团,具有主动靶向作用。
本发明的大分子前药组装后通过氧气氧化交联成纳米颗粒,制备工艺简单。
附图说明
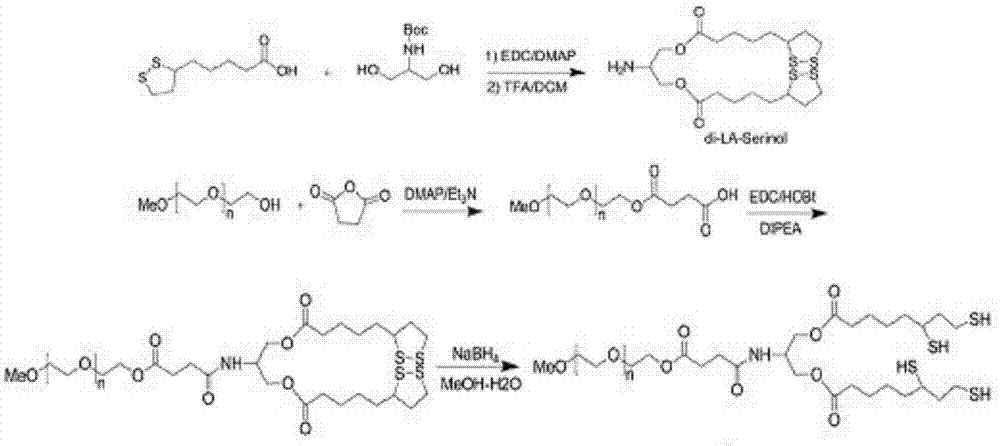
图1.硫辛酸-丝氨醇-聚乙二醇合成路线
图2.双喜树碱-硫辛酸-丝氨醇-聚乙二醇的合成路线
图3.双紫杉醇-硫辛酸-丝氨醇-聚乙二醇的合成路线
图4.双阿霉素-硫辛酸-丝氨醇-聚乙二醇的合成路线
图5.双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇的合成路线
图6.双卡巴他赛-硫辛酸-丝氨醇-聚乙二醇的合成路线
图7.双三尖杉酯碱-硫辛酸-丝氨醇-聚乙二醇的合成路线
图8.双美登素-硫辛酸-丝氨醇-聚乙二醇的合成路线
图9.双依维莫司-硫辛酸-丝氨醇-聚乙二醇的合成路线
图10.双达沙替尼-硫辛酸-丝氨醇-聚乙二醇的合成路线
图11.双达沙替尼-硫辛酸-氨基甘油-聚乙二醇的合成路线
图12.大分子前药中的巯基氧化交联示意图
图13动态光散射法测量双喜树碱-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒尺寸(a)及其透射电镜图(b)
图14.双喜树碱-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒的体外降解
图15.双喜树碱-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒的体外抗肿瘤活性,(a)乳腺癌细胞mcf-7,(b)肝癌细胞hepg-2
图16.双喜树碱-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒的血药浓度(a)与药物在肿瘤和组织中的分布(b)
图17.双喜树碱-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒动物体内抗肿瘤效果(a)和动物体重变化(b)。
具体实施方式
下面结合说明书附图和实施例对本发明的技术方案做进一步详细说明。
本发明的一种大分子前药纳米药物,其特征在于,两个疏水性药物分子由双硫键通过硫辛酸与低分子量聚乙二醇亲水高分子相连形成通式(1)的大分子前药,前药自组装并通过双硫键交联形成内核是含有药物的交联结构、外壳是低分子量聚乙二醇亲水高分子刷的纳米颗粒,尺寸为10~1000纳米。
式中,x是丝氨醇或3-氨基甘油,d是疏水性药物,y是ch2ch2ocoo或ch2ch2oconh,n为6~80。
上述疏水性药物b是紫杉烷类药物包括紫杉醇、多西他赛、卡巴他赛,蒽醌类药物包括阿霉素、表阿霉素、柔红霉素、去甲氧基柔红霉素、阿柔比星、吡柔比星、佐柔比星,喜树碱类药物包括喜树碱、7-乙基-10-羟基喜树碱、羟基喜树碱、拓扑替康、伊立替康、鲁比替康、贝洛替康,三尖杉酯碱类药物包括三尖杉酯碱、异三尖杉酯碱,以及美登素、依维莫司、达沙替尼。
本发明大分子前药纳米药物包括药效学上可接受的载体或助剂,所述助剂是脂肪、磷脂中的一种。
本发明大分子前药纳米药物的制备方法,是通式(1)或(2)的大分子前药在水溶液中组装成直径为10-1000纳米的纳米胶束,加入过氧化氢或在空气中巯基氧化导致分子间交联,形成内核含有疏水药物,外层为亲水性聚乙二醇刷的交联纳米颗粒,经冷冻干燥得到包裹药物的交联纳米颗粒冻干粉。
本发明大分子前药纳米药物在制备抗肿瘤药物中的应用。
本发明药物组合物的优选方案中,药物组合物是液体制剂、固体制剂、半固体制剂、胶囊剂、颗粒剂、凝胶剂、注射剂、缓释制剂或控释制剂。
从体外药理活性等筛选来看,本发明纳米药物表现良好的抗肿瘤等生物活性。试验表明本发明化合物的体内毒性小于原药。因此可作为抗肿瘤等药物用于病人。
本发明制备的纳米药物,跨膜能力强,具有可形成液体制剂、固体制剂、半固体制剂的特性,用于肿瘤等治疗,具有靶向功能;本发明的纳米药物,在体内降解快速释放药物,发挥药效,具有低毒副作用。
本发明的纳米药物在制备抗肿瘤等药物中的应用,将所述大分子前药纳米药物,与药效学上可接受的载体制备成药剂。可将本发明纳米药物与一种或多种固体或液体药物赋形剂和/或辅剂结合,制成可作为人药使用的适当的施用形式或剂量形式。通常本发明药物组合物含有重量百分比0.1-100%的本发明纳米药物。
本发明纳米药物给药途径可为肠道或非肠道,如口服、肌肉、皮下、鼻腔、口腔粘膜、皮肤、腹膜或直肠等。可为注射给药,包括静脉注射、肌肉注射、皮下注射、皮内注射和穴位注射等。给药剂型可以是液体剂型、固体剂型。如液体剂型可以是真溶液类、胶体类、微粒剂型、乳剂剂型、混悬剂型。其他剂型例如片剂、胶囊、滴丸、气雾剂、丸剂、粉剂、溶液剂、混悬剂、乳剂、颗粒剂、栓剂、冻干粉针剂等。
本发明化合物可以制成普通制剂、也可以是缓释制剂、控释制剂、靶向制剂及各种微粒给药系统。
本发明的纳米药物中有二硫键间隔臂或交联键。该间隔臂或交联键在肿瘤组织或细胞内的谷胱甘肽等作用而快速断裂,快速释放原型药物,发挥药效。
本发明纳米药物,提高药物包裹的效率和药物的稳定性,同时使纳米药物易于摄入细胞内,发挥药效。
实施例中合成试剂缩写如下:
四丁基氟化铵三水化合物:tbaf
2-(吡啶基-二硫烷基)乙醇:
1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐:edc·hcl
硫辛酸:la
二异丙基乙基胺:dipea
1-羟基苯并三唑:hobt
4-(二甲基氨基)吡啶:dmap
三光气:btc
叔丁氧羰基:boc
对照例1:
还原型硫辛酸-丝氨醇-聚乙二醇(la-speg)的合成(合成路线见图1)
二硫辛酸-丝氨醇酯(dila-ser)合成:2.97g硫辛酸(la)、3.31g1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc·hcl)加于100ml圆底烧瓶中,加入40mlch2cl2溶解活化30min。将溶于20mlch2cl2的1.25gboc-丝氨醇和2.21g4-(二甲基氨基)吡啶(dmap)加入到上述混合液中,反应12h。反应完成后过滤,将滤液用过量0.1mol/l盐酸充分洗涤,静置后弃去水相。硫酸钠干燥油相,柱层析纯化(淋洗剂:chcl3/ch3oh=20/1,v/v),得到淡黄色油状液体di-boc-la-ser(3.19g,产率86%)。取1gdi-boc-la-ser于10mlch2cl2中,冷至0℃,缓慢滴加0.41g三氟乙酸(tfa),反应2h。将混合物真空浓缩,得到dila-ser粗产物。
还原型硫辛酸-丝氨醇-聚乙二醇(la-speg)的合成:dila-ser粗产物溶解于20ml无水ch2cl2,加入3g甲氧基聚乙二醇琥珀酸单酯(聚乙二醇分子量分别为4000,2000,1000,300),0.42gedc·hcl,0.19g1-羟基苯并三唑(hobt)和0.85g二异丙基乙基胺(dipea),室温反应12h。加入50mlch2cl2稀释,用0.1mhcl洗涤3次。收集合并有机相,无水na2so4干燥,真空浓缩。经硅胶柱色谱(ch2cl2/meoh=20/1,v/v)纯化,得到黄色固体或液体,大约3g。将中间产物溶于15ml甲醇和水混合液中(1/4,v/v),冷却至0℃,加入0.10gnabh4,反应2小时直至溶液变为无色。反应结束后,调ph到3.0,透析纯化低聚物。冻干得到白色固体或粘稠液体还原型硫辛酸-丝氨醇-聚乙二醇(la-speg)各约2.5g,。根据聚乙二醇分子量,产物分别记为la-speg4k,la-speg2k,la-speg1k,la-speg300。
对照例1:
还原型硫辛酸-氨基甘油-聚乙二醇(la-gpeg)的合成
二硫辛酸-氨基甘油酯合成:3g硫辛酸(la)、3.31g1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc·hcl)加于100ml圆底烧瓶中,加入40mlch2cl2溶解活化30min。将溶于20mlch2cl2的1.25gboc-3-氨基甘油和2g4-(二甲基氨基)吡啶(dmap)加入到上述混合液中,反应12h。反应完成后过滤,将滤液用过量0.1mol/l盐酸充分洗涤,静置后弃去水相。硫酸钠干燥油相,柱层析纯化(淋洗剂:chcl3/ch3oh=20/1,v/v),得到淡黄色油状液体。中间产物溶于10mlch2cl2中,冷至0℃,缓慢滴加0.41g三氟乙酸(tfa),反应2h。将混合物真空浓缩,得到二硫辛酸-氨基甘油酯粗产物。
还原型硫辛酸-氨基甘油-聚乙二醇的合成:二硫辛酸-氨基甘油酯粗产物溶解于20ml无水ch2cl2,加入3g甲氧基聚乙二醇琥珀酸单酯(聚乙二醇分子量分别为4000,2000,1000,300),0.42gedc·hcl,0.19g1-羟基苯并三唑(hobt)和0.85g二异丙基乙基胺(dipea),室温反应12h。加入50mlch2cl2稀释,用0.1mhc1洗涤3次。收集合并有机相,无水na2so4干燥,真空浓缩。经硅胶柱色谱(ch2cl2/meoh=20/1,v/v)纯化,得到黄色固体或液体甲氧基聚乙二醇琥珀酰氨基甘油硫辛酸酯,大约3g。将中间产物溶于15ml甲醇和水混合液中(1/4,v/v),冷却至0℃,加入0.10gnabh4,反应2小时直至溶液变为无色。反应结束后,调ph到3.0,透析纯化低聚物。冻干得到白色固体或粘稠液体还原型硫辛酸-氨基甘油-聚乙二醇la-gpeg各约2.5g。根据聚乙二醇分子量,产物分别记为la-gpeg4k,la-gpeg2k,la-gpeg1k,la-gpeg300。
实施例1:
双喜树碱-硫辛酸-丝氨醇-聚乙二醇(dicptla-speg)的合成(合成路线见图2)
0.8g喜树碱(cpt)和0.3g三光气(btc)混合物溶解或悬浮于无水40mlch2cl2中,n2保护下缓慢滴加0.6gdmap的ch2cl2溶液。室温反应2h,然后一次性加入0.4g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤反应混合液体,真空浓缩。硅胶柱色谱纯化粗产物(淋洗剂:etoac/dcm=1/1,v/v),得浅黄色固体喜树碱二硫代吡啶(cpt-ss-pyr)0.6g,产率50%。
分别取对照列1的la-speg4k,la-speg2k,la-speg1k,la-speg300各0.06g,分别加入到0.1gcpt-ss-pyr溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃下搅拌反应48h,通氮气除氧的去离子水中透析12h,冷冻干燥分别得到淡黄色粉末双喜树碱-硫辛酸-丝氨醇-聚乙二醇,并依据聚乙二醇的分子量分别记为dicptla-speg4k、dicptla-speg2k、dicptla-speg1k、dicptla-speg300,各约0.08g。
实施例2:
双紫杉醇-硫辛酸-丝氨醇-聚乙二醇(diptxla-speg)的合成(合成路线见图3)
取0.3g紫杉醇-2’-特丁基二甲基硅烷(ptx-2’-tbdms)和0.12g三光气(btc)混合物溶解或悬浮于无水40mlch2cl2中,n2保护下,缓慢滴加0.29gdmap的ch2cl2溶液,室温反应2h。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。粗产物溶于20ml二氯甲烷中,加入0.5克四丁基氟化铵脱保护。反应液浓缩后,通过硅胶柱色谱纯化(淋洗剂:etoac/dcm=1/1,v/v)。得浅黄色固体紫杉醇二硫代吡啶(ptx-ss-pyr)0.35g。
分别取对照列1的la-speg4k,la-speg2k,la-speg1k,la-speg300各0.06g,分别加入到0.1gptx-ss-pyr溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌48h后,通氮气除氧的去离子水中透析12h。冷冻干燥,各得到白色粉末双紫杉醇-硫辛酸-丝氨醇-聚乙二醇,并依据聚乙二醇的分子量分别记为diptxla-speg4k、diptxla-speg2k、diptxla-speg1k、diptxla-speg300,各约0.07g。
实施例3:
双阿霉素-硫辛酸-丝氨醇-聚乙二醇(didoxla-speg)的合成(合成路线见图4)
0.20g2-(吡啶基-二硫烷基)乙醇溶于无水40mlch2cl2,加入0.12g三光气(btc)混合物中,n2保护下,缓慢滴加0.3gdmap的ch2cl2溶液,室温反应4h。加入0.3g阿霉素盐酸盐(doxhcl)的40mlch2cl2悬浮液,反应4小时。随后,通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v),得到红色固体粉末阿霉素二硫代吡啶(dox-ss-pyr)0.35g。
称取0.06gla-speg1k和0.1gdox-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌48h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到红色固体粉末双阿霉素-硫辛酸-丝氨醇-聚乙二醇(didoxla-speg1k)。
实施例4:
双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇(didoxla-speg)的合成(合成路线见图5)
0.20g2-(吡啶基-二硫烷基)乙醇溶于无水40mlch2cl2,加入0.12g三光气(btc)混合物中,n2保护下,缓慢滴加0.3gdmap的ch2cl2溶液,室温反应4h。加入0.3g阿霉素(dox)的40mlch2cl2悬浮液,反应4小时。随后,通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v),得到红色固体粉末阿霉素二硫代吡啶(dox-ss-pyr)0.35g。
称取0.06gla-speg1k和0.1gdox-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌48h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到红色固体粉末双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇(didoxnla-speg1k)。
实施例5:
双卡巴他赛-硫辛酸-丝氨醇-聚乙二醇的合成(合成路线见图6)
0.3g卡巴他赛(cab)和0.12g三光气混合物溶解或悬浮于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,室温下卡巴他赛的羟基与三光气反应。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。得浅黄色固体阿霉素二硫代吡啶(cab-ss-pyr)0.25g。
称取0.06gla-speg1k和0.1gcab-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌24h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到白色粉末双卡巴他赛-硫辛酸-丝氨醇-聚乙二醇1k(dicabla-speg1k)。
实施例6:
双三尖杉酯碱-硫辛酸-丝氨醇-聚乙二醇的合成(合成路线见图7)
取0.3g三尖杉酯碱(har)和0.12g三光气混合物溶解或悬浮于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,室温下的羟基与三光气反应。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。得浅黄色固体三尖杉酯碱二硫代吡啶(har-ss-pyr)0.25g。
称取0.06gla-speg1k和0.1ghar-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌24h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到白色粉末产物双三尖杉酯碱-硫辛酸-丝氨醇-聚乙二醇1k(diharla-speg1k)。
实施例7:
双美登素-硫辛酸-丝氨醇-聚乙二醇1k的合成(合成路线见图8)
取0.3g美登素(may)溶于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,加入0.20g2,2′-二硫二吡啶,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。得浅黄色固体美登素二硫代吡啶(may-ss-pyr)0.25g。
称取0.06gla-speg1k-和0.1gmay-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌24h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到白色粉末产物双美登素-硫辛酸-丝氨醇-聚乙二醇1k(dimayla-speg1k)。
实施例8:
双依维莫司-硫辛酸-丝氨醇-聚乙二醇(dievela-speg)的合成(合成路线见图9)
取0.3g特丁基二甲基硅氧烷保护的依维莫司(eve)和0.12g三光气混合物溶解或悬浮于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,室温下依维莫司的羟基与三光气反应。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。粗产物溶于20ml二氯甲烷中,加入0.5克四丁基氟化铵脱保护。反应液浓缩后,通过硅胶柱色谱纯化(淋洗剂:etoac/dcm=1/1,v/v)。得浅黄色固体依维莫司二硫代吡啶(eve-ss-pyr)0.25g。
称取0.06gla-speg1k-和0.1geve-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌24h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到白色粉末产物双依维莫司-硫辛酸-丝氨醇-聚乙二醇1k(dievela-speg1k)。
实施例9:
双达沙替尼-硫辛酸-聚乙二醇(didasla-speg)的合成(合成路线见图10)
取0.3g达沙替尼(das)和0.12g三光气混合物溶解或悬浮于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,室温下达沙替尼的羟基与三光气反应。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。得浅黄色固体达沙替尼二硫代吡啶(das-ss-pyr)0.25g。
称取0.06gla-speg1k-和0.1gdas-ss-pyr,共溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃条件下搅拌24h后,通氮气除氧的去离子水中透析12h。冷冻干燥,得到白色粉末产物双达沙替尼-硫辛酸-丝氨醇-聚乙二醇1k(didasla-speg1k)。
实施例10:
双达沙替尼-硫辛酸-氨基甘油-聚乙二醇(didasla-gpeg)的合成(合成路线见图11)
取0.3g达沙替尼(das)和0.12g三光气混合物溶解或悬浮于无水40mlch2cl2中,在n2保护氛围下,缓慢滴加0.29gdmap的ch2cl2溶液,室温下达沙替尼的羟基与三光气反应。随后,一次性加入0.20g2-(吡啶基-二硫烷基)乙醇,搅拌过夜。用去离子水洗涤混合液体系,真空浓缩。通过硅胶柱色谱纯化粗产物(etoac/dcm=1/1,v/v)。得浅黄色固体达沙替尼二硫代吡啶(das-ss-pyr)0.25g。
分别取对照列2的la-gpeg4k,la-gpeg2k,la-gpeg1k,la-gpeg300各0.06g,分别加入到0.1gdas-ss-pyr溶于2ml甲醇/二甲亚砜(2/3,v/v)混合液中,37℃下搅拌反应12h,通氮气除氧的去离子水中透析12h,冷冻干燥分别得到白色粉末双达沙替尼-硫辛酸-氨基甘油-聚乙二醇,并依据聚乙二醇的分子量分别记为didasla-gpeg4k、didasla-gpeg2k、didasla-gpeg1k、didasla-gpeg300,各约0.08g。
实施例12:
大分子前药纳米颗粒的制备
实施例1~11的不同聚乙二醇分子量的大分子前药双喜树碱-硫辛酸-丝氨醇-聚乙二醇(dicptla-speg)、双紫杉醇-硫辛酸-丝氨醇-聚乙二醇(diptxla-speg)、双阿霉素-硫辛酸-丝氨醇-聚乙二醇(didoxla-speg)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇(didoxnla-speg)、双卡巴他赛-硫辛酸-丝氨醇-聚乙二醇(dicbala-speg)、双三尖杉酯碱-硫辛酸-丝氨醇-聚乙二醇(diharla-speg)、双美登素-硫辛酸-丝氨醇-聚乙二醇(dimayla-speg)、双依维莫司-硫辛酸-丝氨醇-聚乙二醇(dievela-speg)、双达沙替尼-硫辛酸-聚乙二醇(didasla-speg)、双达沙替尼-硫辛酸-氨基甘油-聚乙二醇(didasla-gpeg)各0.05g,分别分散于通氮除氧气的10ml水中,慢速搅拌20分钟。通少量氧气,使大分子前药中的巯基氧化交联(交联示意图见图12),得到大分子前药纳米颗粒溶液。冷冻干燥,得到大分子前药纳米颗粒粉末。
动态光散射(dls)方法检测纳米颗粒粒径,结果显示所有各种大分子前药纳米颗粒平均粒径为50~100纳米。透射电镜测量纳米颗粒的形态,结果显示所有大分子前药纳米颗粒均显示球形结构。图13(a)是动态光散射方法测量双喜树碱-硫辛酸-丝氨醇-聚乙二醇1k大分子前药纳米颗粒的平均粒径,其值为75nm,图13(b)是其纳米颗粒形态,呈规整的球形。
实施例13:
双喜树碱-硫辛酸-丝氨醇-聚乙二醇(dicptla-speg)大分子前药纳米颗粒的体外降解
样品:实施例12制备的双喜树碱-硫辛酸-丝氨醇-聚乙二醇1k(dicptla-speg1k)大分子前药纳米颗粒溶于适量的pbs溶液配制成浓度为0.1mmol的溶液。
上述样品分别分成2份,其中一份加入谷胱甘肽(gsh)的pbs缓冲液0.5ml(gsh浓度20mm),另一份加入pbs缓冲液,放置与培养箱中37℃下孵育,用高效液相色谱检测喜树碱含量(agilent1100lc,zorbax反相c18柱,150×4.6mm,5μm,进样量20μl,柱温25℃,检测波长λ=254nm;梯度淋洗:2-90%缓冲液b/a,流速1.0ml/min,缓冲液a:0.1%tfa的去离子水,缓冲液b:0.1%tfa的乙腈)。
结果表明:结果见图14。gsh处理的双喜树碱-硫辛酸-丝氨醇-聚乙二醇(dicptla-speg1k)大分子前药纳米颗粒的pbs溶液在0.5小时后释放出的喜树碱原药达到总量的70%,1小时后释放出的喜树碱原药达到总量的90%;未经gsh处理的大分子前药纳米颗粒溶液1小时后释放出的喜树碱原药仅为总量的10%。
显然,dicptla-speg1k大分子前药纳米颗粒具有gsh敏感性。在gsh作用下,dicptla-speg1k大分子前药纳米颗粒的双硫键断裂,并由断裂后的端基巯基进攻碳酸酯键的羰基,从而快速释放出喜树碱原药。
实施例14:
阿霉素大分子前药纳米颗粒的体外降解
样品:实施例12制备的双阿霉素-硫辛酸-丝氨醇-聚乙二醇1k(didoxla-speg1k)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇1k(didoxnla-speg1k)大分子前药纳米颗粒溶于适量的pbs溶液配制成浓度为0.1mmol的溶液。
上述两种样品分别分成2份,其中一份加入谷胱甘肽(gsh)的pbs缓冲液0.5ml(gsh浓度20mm),另一份加入pbs缓冲液,放置与培养箱中37℃下孵育,用高效液相色谱检测喜树碱含量(agilent1100lc,zorbax反相c18柱,150×4.6mm,5μm,进样量20μl,柱温25℃,检测波长λ=254nm;梯度淋洗:2-90%缓冲液b/a,流速1.0ml/min,缓冲液a:0.1%tfa的去离子水,缓冲液b:0.1%tfa的乙腈)。
结果表明:gsh处理的didoxla-speg1k、didoxnla-speg1k大分子前药纳米颗粒的pbs溶液在0.1小时后释放出的喜树碱原药达到总量的70%,1小时后释放出的阿霉素原药达到总量的85%;未经gsh处理的大分子前药纳米颗粒溶液1小时后释放出的阿霉素原药低于总量的10%。
显然,didoxla-speg1k、didoxnla-speg1k大分子前药纳米颗粒具有gsh敏感性。在gsh作用下,didoxla-speg1k、didoxnla-speg1k大分子前药纳米颗粒的双硫键断裂,并由断裂后的端基巯基进攻碳酸酯键或氨基甲酸酯键的羰基,从而快速释放出阿霉素原药。
实验例15:
药理实验:mtt法癌细胞活力试验
双喜树碱-硫辛酸-丝氨醇-聚乙二醇1k(dicptla-speg1k)大分子前药纳米颗粒的抗肿瘤活性
mcf-7人乳腺癌细胞、肝癌细胞hepg2以8×103个/孔的接种量接种于96孔培养板,5%co2、37℃培养箱中培养24h后,每孔加入不同浓度的实施例12的dicptla-speg1k大分子前药纳米颗粒溶液、对照喜树碱药物溶液(喜树碱溶于生理盐水,用dmso增溶)各100μl,使最终筛选的药物终浓度分别为1、2、5、10、20μg/ml,继续培养24h后;分别加入50μlmtt温育4h,弃去培养基,加入150μldmso,在平板摇床上摇匀,酶标仪495nm读板,根据测得的吸光度值计算细胞抑制率。数据表示为平均数±sd(n=6)。
结果如图15所示,dicptla-speg1k大分子前药纳米颗粒对乳腺癌细胞mcf-7及肝癌细胞hepg-2具有良好增长抑制与杀伤作用,保留了喜树碱强抗肿瘤活性,其对应两种肿瘤细胞的ic50值分别为2.06μg/ml和1.89μg/ml,且细胞毒性随着药物浓度的增加而增强,具有浓度依赖性。在相同药物剂量下,dicptla-speg1k大分子前药纳米颗粒表现出细胞毒性较游离态的喜树碱原药弱。这是因为双喜树碱-硫辛酸-聚乙二醇1k大分子前药纳米颗粒在pbs(ph7.4)中自组装为稳定的纳米颗粒,相比游离态的喜树碱药物释放,可能具有一定缓释作用。因此dicptla-speg1k大分子前药纳米颗粒在24h内表现出相对游离态药物较低的抗肿瘤活性。
实验例16:
药理实验:mtt法癌细胞活力试验
阿霉素大分子前药纳米颗粒的抗肿瘤活性
mcf-7人乳腺癌细胞、肝癌细胞hepg2以8×103个/孔的接种量接种于96孔培养板,5%co2、37℃培养箱中培养24h后,每孔加入不同浓度的实施例12的双阿霉素-硫辛酸-丝氨醇-聚乙二醇1k(didoxla-speg1k)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇1k(didoxnla-speg1k)大分子前药纳米颗粒溶液、对照阿霉素溶液(阿霉素溶于生理盐水,用dmso增溶)各100μl,使最终筛选的药物终浓度分别为1、2、5、10、20μg/ml,继续培养24h后;分别加入50μlmtt温育4h,弃去培养基,加入150μldmso,在平板摇床上摇匀,酶标仪495nm读板,根据测得的吸光度值计算细胞抑制率。数据表示为平均数±sd(n=6)。
结果表明,双阿霉素-硫辛酸-丝氨醇-聚乙二醇1k(didoxla-speg1k)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇1k(didoxnla-speg1k)大分子前药纳米颗粒对乳腺癌细胞mcf-7及肝癌细胞hepg-2具有良好增长抑制与杀伤作用,保留了阿霉素强抗肿瘤活性,其对应两种肿瘤细胞的ic50值与阿霉素相近,且细胞毒性随着药物浓度的增加而增强,具有浓度依赖性。在相同药物剂量下,大分子前药纳米颗粒表现出细胞毒性较游离态的阿霉素原药弱。这是因为大分子前药纳米颗粒在pbs(ph7.4)中自组装为稳定的纳米颗粒,相比游离态的阿霉素药物释放,可能具有一定缓释作用。因此didoxla-speg1k、didoxnla-speg1k大分子前药纳米颗粒在48h内表现出相对游离态药物较低的抗肿瘤活性。
实施例17:
双喜树碱-硫辛酸-丝氨醇-聚乙二醇1k(dicptla-speg1k)大分子前药纳米颗粒的药代动力学研究
将种植肿瘤尺寸为100-150mm3mcf-7的雌性balb/c裸鼠,随机分成伊立替康和dicptla-speg1k大分子前药纳米颗粒组(n=3)。通过尾静脉注射5mgcpt/kg伊立替康和dicptla-speg1k大分子前药纳米颗粒溶液,分别在0.5h,1h,2h,4h,6h,8h和24h,收集血液样品,并以3,000rpm离心10min以获得血浆。然后,利用乙腈提取血浆中伊立替康和未水解的dicptla-speg1k大分子前药的量。具体步骤:将100μl血浆样品于2ml离心管中,加入1ml含有0.5%乙酸的乙腈,混合均匀。混合物经11,000rpm离心10分钟后,将获得的上清液进行hplc分析。
为了评估药物制剂的生物体内组织分布,荷载mcf-7肿瘤的balb/c裸鼠在注射5mgcpt/kg伊立替康和dicptla-speg1k大分子前药纳米颗粒溶液0.5h,2h和6h后,通过颈椎脱位方法处死裸鼠,切除包括心脏,肝脏,脾脏,肺脏,肾脏和肿瘤在内的主要器官,用0.9%生理盐水漂洗并称重,均质化。使用与血浆提取的相同程序从组织匀浆中提取cpt,hplc测定药物浓度,并相应地计算组织中相应的cpt含量。
结果见图13(a)。在给药后4-8小时内,游离伊立替康能够明显地从血液循环中清除。该结果与dicptla-speg1k大分子前药纳米颗粒形成显著对比,延长了药物的血液循环时。这是因为,dicptla-speg1k大分子前药纳米颗粒有利于避免网状内皮系统(res)的快速摄取。第二,dicptla-speg1k大分子前药保持其良好的稳定性而不会在生理环境中分解。第三,peg的存在将进一步延长血液循环。而且,短链peg不易导致机体产生peg抗体,从而降低了药物的血浆消除速率。
纳米颗粒在移植mcf-7肿瘤的balb/c裸鼠肿瘤和其他器官中的分布见图16(b)。可见,dicptla-speg1k大分子前药纳米颗粒主要在2小时后分布到网状内皮系统(res)器官,例如肺,脾,特别是肝脏,而相应的伊立替康水平较低。dicptla-speg1k大分子前药纳米颗粒在这些器官中的较大累积可能是由于制剂的纳米级特征,其可以提供长期治疗效果。更重要的是,用dicptla-speg1k1k大分子前药纳米颗粒处理的实验组在肿瘤中显示出比伊立替康治疗组更高的cpt浓度(p<0.01)。可见,dicptla-speg1k大分子前药纳米颗粒可以减少res系统的快速捕获并且由于epr效应而被动地积聚在肿瘤中。因此,与单独的伊立替康相比,由dicptla-speg1k大分子前药纳米颗粒可延长血浆半衰期并显着增强体内向肿瘤组织的分布。
实验例18:
双喜树碱-硫辛酸-丝氨醇-聚乙二醇1k(dicptla-speg1k)大分子前药纳米颗粒体内药效和毒性实验
裸鼠模型的制备:收集培养的hepg2细胞悬液,浓度为1×107个/ml,以每只0.1ml接种于裸小鼠右侧腋窝皮下。分组与给药:裸鼠移植瘤用游标卡尺测量移植瘤直径,肿瘤生长至75mm3时将动物随机分组。同时,各组裸鼠开始给药,给药方案见组别与给药方案,使用测量瘤径的方法,动态观察受试样品的抗肿瘤效应。肿瘤体积(tv)的计算公式为:tv=1/2×a×b2(式3-2),其中a、b分别表示长宽。
组别及给药方案:空白组:生理盐水,静脉注射,每三天注射一次,体积为0.2ml,连续3周。
对照组:伊立替康溶于生理盐水(微量dmso助溶,给药量10mg/kg),静脉注射,每三天注射一次,体积为0.2ml,连续3周。
药物组:实施例12的dicptla-speg1k大分子前药纳米颗粒溶液(给药量相当于喜树碱10mg/kg),静脉注射,每三天注射一次,体积为0.2ml,连续3周。期间,测量体重变化。
抗肿瘤活性和体重变化结果见图17。从抗肿瘤活性(图17a)来看,本发明dicptla-speg1k大分子前药纳米颗粒具有很好的抑制肿瘤生长作用,且动物体重没有下降(图14b),显示无毒性。
实验例19:
双阿霉素-硫辛酸-丝氨醇-聚乙二醇大分子前药纳米颗粒体内药效和毒性实验
裸鼠模型的制备:收集培养的hepg2细胞悬液,浓度为1×107个/ml,以每只0.1ml接种于裸小鼠右侧腋窝皮下。分组与给药:裸鼠移植瘤用游标卡尺测量移植瘤直径,肿瘤生长至75mm3时将动物随机分组。同时,各组裸鼠开始给药,给药方案见组别与给药方案,使用测量瘤径的方法,动态观察受试样品的抗肿瘤效应。肿瘤体积(tv)的计算公式为:tv=1/2×a×b2(式3-2),其中a、b分别表示长宽。
组别及给药方案:空白组:生理盐水,静脉注射,每三天注射一次,体积为0.2ml,连续3周。
对照组:阿霉素溶于生理盐水(微量dmso助溶,给药量10mg/kg),静脉注射,每三天注射一次,体积为0.2ml,连续3周。
药物组:实施例12的双阿霉素-硫辛酸-丝氨醇-聚乙二醇1k(didoxla-speg1k)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇1k(didoxnla-speg1k)大分子前药纳米颗粒溶液(给药量相当于喜树碱10mg/kg),静脉注射,每三天注射一次,体积为0.2ml,连续3周。期间,测量体重变化。
结果表明,本发明双阿霉素-硫辛酸-丝氨醇-聚乙二醇1k(didoxla-speg1k)、双阿霉素-n-硫辛酸-丝氨醇-聚乙二醇1k(didoxnla-speg1k)大分子前药纳米颗粒给药的动物肿瘤体积远小于阿霉素对照组,显示大分子前药纳米颗粒具有很好的抑制肿瘤生长作用;纳米颗粒给药组动物体重没有下降(图14b),显示毒性低于阿霉素;动物心肌免疫组化显示,大分子前药纳米颗粒组动物心肌组织无明显变化,阿霉素组动物心肌有显著组织学改变。
实施例20:
大分子前药纳米颗粒的制备
实施例1、2、3的不同聚乙二醇分子量的大分子前药双喜树碱-硫辛酸-丝氨醇-聚乙二醇(dicptla-speg)、双紫杉醇-硫辛酸-丝氨醇-聚乙二醇(diptxla-speg)、双阿霉素-硫辛酸-丝氨醇-聚乙二醇(didoxla-speg)各0.05g,分别加入蛋黄卵磷脂、脂肪酸甘油酯或二硬脂酰磷酸胆碱0.05克,分散于通氮除氧气的10ml水中,慢速搅拌20分钟。通少量氧气,使大分子前药中的巯基氧化交联(交联示意图见图12),得到3类大分子前药纳米颗粒溶液。冷冻干燥,得到3类大分子前药纳米颗粒粉末。动态光散射(dls)方法检测纳米颗粒粒径,结果显示所有各种大分子前药纳米颗粒平均粒径为50~300纳米。
- 还没有人留言评论。精彩留言会获得点赞!