一种由醛糖二酸制备粘康酸和呋喃的方法与流程
本发明涉及通过选择性催化脱羟基反应由醛糖二酸制备糖酸平台化学品。特别地,本发明涉及粘康酸和呋喃的制备,所述粘康酸和呋喃是制备各种各样的工业上重要的化学品和药物结构单元的重要中间体。
背景技术:
最近,纤维素生物质作为化学品和燃料的可再生原料备受关注。工业相关的化合物通常是通过原油衍生工艺或采用生物技术例如发酵来制备。该领域一个主要的挑战是生物化合物通常太过富氧以至于不能与当前的石油基工业相容。对有效脱氧(deoxygenation)方法的研究导致对催化脱氧脱水(deoxydehydration)(DODH)方法的日益增长的兴趣,以便将生物基资源选择性地转化为目标化学品。
在现有技术中,Rennovia(WO 2010/144862 A2)描述了一种将葡萄糖转化为己二酸产品的方法,通过催化氧化反应(葡萄糖转化为葡糖二酸)和通过使用加氢脱氧催化剂、卤素源和H2的催化加氢脱氧反应(catalytic hydrodeoxygenation)(葡糖二酸转化为己二酸)。
Shiramizu和Toste(2013)描述了通过使用DODH和加氢反应将粘酸转化为己二酸酯的适度的转化(43-94%)。根据Shiramizu和Toste的粘康酸制备中的缺点之一是所用的3-戊醇或1-丁醇的显著的化学计量的牺牲(每摩尔的粘康酸产品耗费2-4摩尔醇)。因此,粘康酸制备中的缺点之一是所用的3-戊醇或1-丁醇的显著的化学计量的牺牲。粘康酸的合成使用高温(155℃)。该方法还使用化石戊醇代替可再生醇,例如甲醇。此外,作为还原剂的戊醇产生了4摩尔当量的作为副产物的氧化戊醇。
Li等人(2014)描述了通过DODH反应将粘酸转化为粘康酸的甚至更有效的转化(高达99%),其通过氧化铼(oxorhenium)复合物来催化。通过结合DODH和转移氢化反应(transfer-hydrogenation reaction),粘酸可以被成功地转化为己二酸。Li等人在他们的反应中除了催化剂外使用了化石戊醇和酸。Shiramizu&Toste和Li等人都描述了甲基三氧化铼(methyltrioxorhenium)作为催化剂和3-戊醇(或1-丁醇)作为还原剂的使用,反应温度从120℃至170℃变化。
呋喃化学品也可以通过醛糖二酸的脱羟基反应制备。然而,当前的方法使用强无机酸作为试剂,并且反应时间长,长达40小时(FR2723945,Taguchi et al.,2008)。
Ahmad等人(2011)描述了通过亚硫酸盐驱动的氧化铼催化的乙二醇的脱氧脱水反应制备烯烃。使用固体还原剂,其产生固体废物材料。氢被提及为一种经济上可行的还原剂,但是这仅用四氢呋喃示出。Ahmad等人没有描述通过使用相同的方法设置并且仅改变温度能够制备呋喃或粘康酸的方法。
众所周知,原油是有限但必不可少的资源。另一方面,醛糖二酸,例如半乳糖酸,可以由果胶和其它不可食用的碳水化合物产生。通过将醛糖二酸转化成粘康酸和/或呋喃,打开了允许从生物基资源制备多种化合物的一扇门,否则其将由原油原料制备。因此,需要一种避免有限资源的使用并且替代地使用有利的技术,利用有机合成和催化剂工具并且允许所需的化学品的容易的可扩展性、选择性和即时纯化的环保方法。
技术实现要素:
本发明的目的是通过环境友好的方法由醛糖二酸制备粘康酸和呋喃化学品。
此外,本发明描述了通过使用有机合成和催化工具、特别是催化脱羟基反应将醛糖二酸例如半乳糖二酸和葡糖二酸转化为粘康酸和呋喃的选择性方法。
更确切地说,本发明的方法的特征在于权利要求1的特征部分中公开的内容。本发明的用途的特征在于权利要求17和18。
本发明的优点包括将不可食用的碳水化合物转化为中间体,其可用于工业上重要的化学品和药物结构单元的制备。此外,本发明所述的方法允许所获得的产物的容易的可扩展性和纯化。
另一个优点是该方法是环保的,并且带来低能量消耗(基于例如反应温度和时间)和低废物产生(基于例如H2还原剂)。
下面参考某些实施例更密切地描述本发明。
附图说明
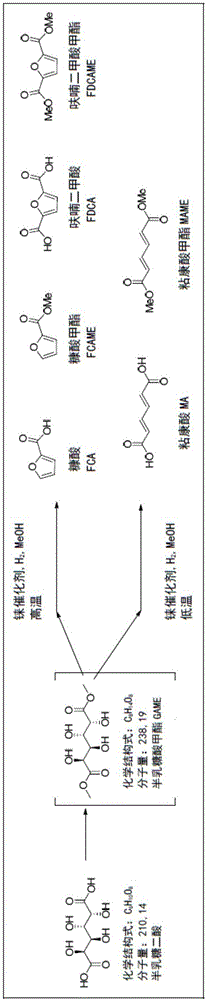
图1描述了通过铼催化的脱羟基反应的从半乳糖二酸到粘康酸和呋喃的两种温度控制路线。
具体实施方式
本发明涉及糖酸平台化学品的制备,更准确地是通过选择性催化脱羟基反应从醛糖二酸到粘康酸和呋喃例如糠酸和呋喃二甲酸的制备。
醛糖二酸是糖酸族,其中糖的末端羟基已被末端羧酸取代,并且其具有化学式HOOC-(CHOH)n-COOH的特征。醛糖二酸的命名法是基于它们衍生的糖。例如,葡萄糖被氧化成葡糖二酸,半乳糖被氧化成半乳糖酸和木糖被氧化成木糖酸。与其母体糖不同,醛糖二酸在其碳链的两端具有相同的官能团。因此,两种不同的醛糖二酸可制备相同的粘康酸。
根据本发明的一个优选实施方式,所述方法包括在过渡金属催化剂的存在下,于加压容器中将醛糖二酸、溶剂和还原剂加热至90至300℃的温度反应预定的反应时间并纯化所得的一种或多种产物。
特别地,所述方法包括醛糖二酸的选择性催化脱羟基反应,其中催化选择性地指向粘康酸通过使用90至150℃的催化温度,例如100至120℃,优选约100℃。另一方面,所述催化可以选择性地指向呋喃通过使用150至300℃的催化温度,例如150至250℃,优选约200℃。然而,所有II至IX的化合物也能在120至150℃的温度下制备。反应温度可以基于目标产物进行选择和调节,如在下面的实施例中所描述的。反应通常从半乳糖酸开始,但也可以使用其他的醛糖二酸,例如葡糖二酸。
如前所述,本发明的催化可以仅通过调节反应温度和时间选择性地指向粘康酸路线或呋喃路线。这种令人惊讶的和有利的发现在本领域中尚未公开。
本发明一个重要的方面是选择催化剂、溶剂和还原剂的有效的和功能的组合。现有技术的早期尝试不能促进将低级(即短)醇如甲醇、乙醇和正丁醇用于还原步骤。本发明的发明人已经设法开发向所需的终产品提供优异结果的组合。因此,这种组合的一个实例是使用甲基三氧化铼催化剂以及低级醇如甲醇作为溶剂以及氢气作为还原剂。当使用半乳糖二酸作为醛糖二酸并且如本文所述的催化剂/还原剂/溶剂组合时,从粘康酸路线获得的产物包含粘康酸(MA)和粘康酸甲酯(MAME)。因此,呋喃路线提供了诸如糠酸(FCA)、糠酸甲酯(FCAME)、呋喃二甲酸(FDCA)和呋喃二甲酸甲酯(FDCAME)的产物。
然而,也可以使用除铼之外的其它过渡金属催化剂,例如钼、钒和钯催化剂。
使用上述组合的一个主要优点是氢带来作为副产物的H2O,因此仅留下醇溶剂,例如甲醇,其在纯化步骤中易于洗涤或蒸馏。氢也可以再循环并且比其它现有技术的还原剂例如1-丁醇更便宜。除了醇之外的其它还原剂在纯化步骤中也是有问题的,并且必须物理除去。因此,该方法特别地环保并且仅产生少量的废物。
与能量消耗保持低的现有技术方法相比,本发明所述的方法还能够在更低的温度下进行。关于前述,反应时间设定为1分钟至70小时,优选1至2小时,特别是1至60分钟,使得该方法更加能量有效。然而,反应时间取决于目标产物。通常更长的时间(例如48至70小时)对于获得MA/MAME是必需的,较短的时间(1分钟至60分钟,优选1分钟至10分钟)可适用于制备呋喃产品。
所制备的产物的纯化包括过滤任何固体沉淀物,用醇洗涤沉淀物并干燥洗涤的产物,例如通过蒸发。随后蒸发含有本发明所需产物的有机相,然后通过例如二氧化硅柱色谱法纯化。结果通过本领域通常公知的进一步分析方法证实。
通过以下非限制性实施例说明本发明。然而,应当理解,以上的描述和实施例中给出的实施方式仅用于说明目的,并且在权利要求的范围内可以进行各种改变和修改。
实施例
以下实施例的相关化合物的编号和结构式:
半乳糖二酸(I)
2,4-己二酸(2E,4E)(II)
2,4-己二酸1,6-二甲酯(2E,4E)(III)
2-呋喃甲酸(IV)
2-呋喃甲酸甲酯(V)
2,5-呋喃二甲酸(VI)
2,5-呋喃二甲酸2,5-二甲酯(VII)
2,4-己二酸1,6-二乙酯(2E,4E)(VIII)
2,4-己二酸1,6-二丁酯(2E,4E)(IX)
通用方法
反应在25ml聚四氟乙烯涂覆的压力容器中进行。使用对每种产物化合物进行外部校准的GC-FID测定产物产率。使用相应羧酸的标准酯化方法产生用于校准的酯标准。使用GC-MS和NMR分析证实结果。
实施例1 用于化合物II-VII的制备的半乳糖二酸(I)的催化脱羟基反应
将半乳糖二酸(1.0g,4.76mmol)、甲基三氧化铼(0.12g,0.47mmol,10mol%)和甲醇(10ml)装入反应容器中。将反应容器用氢气加压并加热至反应温度(表1)。在指定的反应时间后,将混合物冷却至室温,过滤任何固体沉淀,用甲醇(5ml)洗涤并干燥。在旋转蒸发器中浓缩溶剂级分。通过快速硅胶柱色谱法纯化。使用GC-FID、GC-MS和NMR分析不同的级分。
表1.半乳糖二酸和MTO/MeOH的实验
实施例2 用于化合物VIII的制备的半乳糖二酸(I)的催化脱羟基反应
将半乳糖二酸(1.0g,4.76mmol),甲基三氧化铼(0.12g,0.47mmol,10mol%)和乙醇(10ml)装入反应容器中。将反应容器用氢气加压并加热至反应温度(表2)。在指定的反应时间后,将混合物冷却至室温,过滤任何固体沉淀,用甲醇(5ml)洗涤并干燥。在旋转蒸发器中浓缩溶剂级分。使用GC-FID和GC-MS分析不同的级分。
表2.半乳糖二酸和MTO/MeOH的实验
实施例3用于化合物IX的制备的半乳糖二酸(I)的催化脱羟基反应
将半乳糖二酸(1.0g,4.76mmol),甲基三氧化铼(0.12g,0.47mmol,10mol%)和1-丁醇(10ml)装入反应容器中。用氢气加压反应容器并加热至反应温度(表3)。在指定的反应时间后,将混合物冷却至室温,过滤任何固体沉淀,用丁醇(5ml)洗涤并干燥。在旋转蒸发器中浓缩溶剂级分。使用GC-FID和GC-MS分析不同的级分。
表3.半乳糖二酸和MTO/MeOH的实验
实施例4来自反应#1的粘酸甲酯的纯化
浓缩溶剂级分后得到紫色粉末(528mg),将其溶于丙酮(10g)中,然后加入硅胶(1g)中。然后将其蒸发成粉末,并用溶剂10%乙酸乙酯/90%己烷从闪蒸柱(11cm硅胶)洗脱。从产物级分中除去溶剂,得到化合物III,为白色粉末,110.4mg,产率8.2%。1H NMR(D6-DMSO)3.70(6H,s,CH3)、6.49(2H,d,J 13.95烯烃H)、7.40(2H,d,J 13.95,烯烃H)。13C NMR(D6-DMSO)165、141、128、51。GC-MS m/z 170。
引文列表–专利文献
1.WO 2010/144862 A2
2.FR2723945
引文列表–非专利文献
1.Shiramizu,M.and Toste,F.D.,2013,Expanding the Scope of Biomass-derived Chemicals through Tandem Reactions Based on Oxorhenium-Catalyzed Deoxydehydration,Angew.Chem.Int.,Vol 52,pp.12905-12909,DOI:10.1002/anie.201307564.
2.Li,X.,Wu,D.,Lu,T.,Yi,G.,Su,H.and Zhang,Y.,2014,Highly Efficient Chemical Process to Convert Mucic Acid into Adipic Acid and DFT Studies of the Mechanism of the Rhenium-Catalyzed Deoxydehydration,Angew.Chem.,Vol 126,pp.1-6,DOI:10/1002/ange.201310991.
3.Taguchi,Y.,Oishi,A.,Iida,H.,2008,One-step Synthesis of Dibutyl Furandicarboxylates from Galactaric Acid,Chem.Lett.,Vol 37,pp.50-51,DOI:10.1246/cl.2008.50.
4.Ahmad,I.,Chapman,G.,Nicholas,K.M.,2011,Sulfite-Driven,Oxorhenium-Catalyzed Dehydroxylation of Glycols,Organometallics,Vol 30,pp.2810-2818.
- 还没有人留言评论。精彩留言会获得点赞!