用于有机电致发光元件的有机铱配合物的制作方法
本发明涉及一种提供适合作为有机电致发光(EL)元件的有机铱配合物的技术,特别是涉及一种适合作为绿色至黄色的发光材料的有机铱配合物。
背景技术:
有机电致发光(Electroluminescence,EL)元件作为下一代显示器或照明的技术开发备受期待。其特征为:具有消耗电力少,可薄型化,反应速度优异,在暗处与明处均可清晰地显示影像等优点。
作为该有机EL元件的基本结构,以一对电极夹住一层或多层有机化合物的三明治结构是适用的。具体而言,有人提出以阴极/电子输送层/发光层/空穴输送层/阳极/玻璃基板这样的三明治结构为主要构成,并为了进一步提高特性而适当追加空穴(电子)注入层、缓冲层、层间绝缘膜等的结构的元件。在位于三明治结构中心的发光层中使用各种发光材料,其特性为:寻求使从阴极或阳极输送的电子或空穴容易流动、发光效率优异、具有耐久性等。
由于这样的要求特性,相对于以往适用的荧光材料,目前寻求开发磷光材料来作为用于有机EL元件的发光材料。在有机EL元件中,因为激发单重态与激发三重态产生激发分子的机率为1:3,故相对于通过从激发单重态回到基态而发光的荧光材料,通过从激发三重态状态回到基态而显示磷光的磷光材料更受到瞩目。已经有人开发各种有机金属配合物作为这种磷光材料,例如,有人提出如下式所示的有机金属配合物,其为将具有杂环且具备C-N结构的配位基(C-N配位基)与β-二酮等的配位基配位于铂或铱等金属原子而成。具体而言,专利文献1公开了一种包含具备两个苯环的配位基(二苯基二酮)作为β-二酮配位基的有机铱配合物(专利文献1的S02等)。另外,专利文献2公开了一种包含具有两个经丁氧基取代的苯环的配位基(四-丁氧基二苯基二酮)作为β-二酮配位基的有机铂配合物(专利文献2的式[1-1])。以上专利文献所记载的有机金属配合物中,通过应用具有苯环的配位基作为β-二酮配位基,从而谋求发光效率的提高。
[化1]
[现有技术文献]
[专利文献]
专利文献1:日本特开2005-35902号公报
专利文献2:日本特开2008-222635号公报
技术实现要素:
发明所要解决的课题
顺便提及,作为有机EL元件的发光效率的评价基准,已知有“电流效率(cd/A)”与“量子效率(%)”。电流效率表示相对于单位电流量的亮度(或考虑视感度的光强度),另一方面,量子效率为作为光能可取出的光子数相对于消耗功率(注入的载子数)的比率。量子效率可排除在消耗功率当中无法释出为光能的部分(例如电阻所导致的损耗量)。因此,相较于电流效率,量子效率可以说是能够接近有机EL元件的实际发光效率的评价。在此背景下,若从专利文献1、2记载的有机金属配合物来看,虽然是针对作为发光效率的电流效率高的有机金属配合物来进行研究,但其并非一定具有高量子效率。
另外,上述量子效率会在薄膜等高分子(固体)中有效地显示出较高的数值。这是因为一般在安装到有机EL元件时,有机金属配合物并非掺入到溶液中或溶剂中,而是掺入到高分子薄膜中,从而应用于发光层。关于这一点,若从专利文献1、2等所记载的有机金属配合物来看,虽然其在有机溶剂等溶液中显示出一定程度的量子效率,但其在薄膜等高分子(固体)中,量子效率大多较低,不是在薄膜中显示出高量子效率的配合物。
于是,本发明的目的在于提供一种在(高分子)薄膜中也具有高量子效率的有机金属配合物来作为用于有机EL元件的发光材料,特别是关于绿色至黄色的电致发光,提供一种可制作高量子效率的有机EL元件的有机金属配合物。
用于解决课题的手段
为了解决上述课题,本申请发明人等着眼于具有以铱作为中心原子的有机铱配合物。作为有机金属配合物,虽然如专利文献2所述那样开发出一种铂配合物,但铂配合物的平面性高,作为中心元素的铂原子的配位基具有空位,从而容易发生能量损耗。具体而言,其受到下述各种相互作用的影响:缔合(会合)·准分子(エキシマー)形成等的分子间相互作用(所谓的自我组织)、或与溶剂·基质(母材)等介质的相互作用、或更进一步与共存的其他分子的缔合等。相对于此,有机铱配合物中的三个配位基形成立体的构象,不会发生上述铂配合物般的各种相互作用,而不易产生能量损耗,故本发明人认为其容易成为量子效率高的材料。
再者,关于量子效率,着眼于决定量子效率的因素之一的发光材料的“发光(PL)量子产率”。如下式所示,在将量子效率大致分为“外部量子效率”与“内部量子效率”的情况下,此PL量子产率为决定内部量子效率的因素之一。发光材料追求高内部量子效率,作为内部量子效率的决定因素,“激子产生效率”及“PL量子产率”产生的影响特别大。其中“激子产生效率”取决于荧光材料或磷光材料的差别,故为了提高内部量子效率,高PL量子产率变得重要。另外,下式中的载子平衡(キャリアバランス)为由材料的组合或膜厚控制等的元件结构所决定的因素。
[量子效率]
外部量子效率=(光萃取效率)×内部量子效率
内部量子效率=(激子产生效率)×(PL量子产率)×(载子平衡)
于是,本申请发明人等特别针对在高分子薄膜中显示高发光(PL)量子产率的有机铱配合物进行深入研究。接着,着眼于在高分子薄膜中产生与溶液溶剂中不同的发光色、即所谓的“刚致变色(リジッドクロミズム)”的有机铱配合物,发现在产生该刚致变色的配合物中,存在有在高分子薄膜中显示高发光量子产率的配合物,从而完成下述本发明。此处,“变色(クロミズム)”指的是物质的光学特性因来自外部的刺激而出现可逆性变化的现象。其中“刚致变色”指的是,诱发变色的外部刺激取决于介质分子的种类,而发光色会根据介质分子是溶液还是固体而变化。由于以上原因,本申请发明人等得出了以下的本发明,并作为产生刚致变色的有机铱配合物。
即,本发明涉及一种由下式所示的、用于有机电致发光元件的有机铱配合物,其通过将由两个原子团(A1、A2)所构成的C-N配位基与具有由叔丁基取代的两个苯基的线对称β-二酮配位基配位于铱原子而形成。
[化2]
(上式中,R1、R2、R3为叔丁基或氢原子,并至少具有一个以上的叔丁基;在具有两个叔丁基的情况下,它们也可互相键合而形成饱和烃环;A1、A2为不饱和烃环,至少一个为单环,且至少一个为杂环)。
本发明的第一特征为:以具有苯基且呈现线对称结构作为配位基的β-二酮为前提,同时具有叔丁基作为取代基。若是具有这种β-二酮的有机铱配合物,则容易成为高发光量子产率的配合物。然而,即使是这种具有叔丁基的有机铱配合物,在周围的介质分子为固体状高分子薄膜的情况下,也未必成为高量子产率的配合物。因此,本申请发明人针对在高分子薄膜中也具有高发光量子产率的有机铱配合物进行进一步研究,结果想到了采用下述结构作为C-N配位基的本发明。相对于此,先前技术在设计有机金属配合物的结构时,对于C-N配位基,主要是从波长位移方面考虑,只要是可呈现目标发光色(红、蓝、绿等),即可从大量列举的结构中任意选择。即,先前技术几乎没有限定C-N配位基,而仅以β-二酮的结构为特征结构。
如上所述,本发明采用上述β-二酮配位基,同时将C-N配位基限定于下列结构。如此,作为在提供于高分子薄膜中也具有高发光量子产率的有机铱配合物时,将C-N配位基的结构特定化的理由,本申请发明人等认为,对于在薄膜中显示高发光量子产率的铱配合物的发光激发状态,其基于从O-O配位基到C-N配位基的电荷移动迁移,且C-N配位基优选为电子接受性较高的结构。此处,在铱配合物于基态与激发状态下的极性不同时,因周边介质分子的影响而使能级(带隙)ΔE产生变化成为刚致变色的主要原因。接着,在通过上述电子接受性的C-N配位基作出电荷移动型的激发状态的情况下,可能会出现刚致变色。其结果是,认为可得到在薄膜中随着ΔE变化的发光量子产率较高的有机铱配合物。
由于以上原因,本申请发明人等不仅限定β-二酮配位基的特征,也使C-N配位基的种类为特定结构,而由此想到本发明:特别在薄膜中具有高发光量子产率的有机铱配合物。作为本发明的有机铱配合物中所产生的刚致变色,对于发光光谱的峰值,相较于在有机溶剂中的波长(λPL),高分子薄膜中的波长(λPL)会更加偏移至短波长侧。这种刚致变色所导致的波长位移(ΔλPL)为15nm以上100nm以下时,特别容易成为高发光量子效率的有机铱配合物。另外,ΔλPL特别优选为25nm以上60nm以下。
以下对本发明的有机铱配合物进行详细说明。
本发明的有机铱配合物通过将β-二酮及两个C-N配位基配位于3价的铱原子而得到。两个C-N配位基为彼此相同的结构,β-二酮配位基具有线对称的结构。以下说明C-N配位基及β-二酮配位基的具体结构。
在本发明中所应用的β-二酮配位基如下式所示,具有两个经叔丁基取代的苯基。
[化3]
上式中,R1、R2、R3为叔丁基或氢原子。一个苯基至少具有一个以上的叔丁基,优选具有两个以上的叔丁基。两个叔丁基可互相键合而形成饱和烃环。
以下显示特别优选的β-二酮配位基的结构。下式中,t-Bu表示叔丁基。
[化4]
接着说明C-N配位基。本发明所应用的C-N配位基的通式如下式所示。
[化5]
在上述C-N配位基中,上侧的原子团A1为不饱和烃环,优选为6元环。A1优选为单环的杂环或单环的苯环,杂环中的杂原子优选为氮。另外,A1的侧链可具有任意的取代基,取代基优选为氟原子(F)、三氟(-CF3)基或氰基(-CN)。
具体而言,原子团A1优选为下式所示的结构中的任一种。在下式中,A1与A2的键合位置以向下的虚线表示。
[化6]
C-N配位基下侧的原子团A2优选为单环的杂环,杂原子优选为氮。另外,A2的侧链可具有任意的取代基,取代基优选为氟原子(F)、三氟(-CF3)基、烷基(-R,碳原子数为1以上10以下,优选为1以上4以下)、烷氧基(-OR,碳原子数为1以上4以下)。此外,与A1相同,A2也为不饱和烃,优选为6元环。
具体而言,原子团A2优选为下式所示的结构中的任一种。在下式中,A2与A1的键合位置以向上的虚线表示。
[化7]
在上式中,R是碳原子数为1以上10以下的烷基。
具有以上所说明的原子团A1及A2的C-N配位基可列举如下的例子。
[化8]
式中,*符号表示与铱键合的碳。
[化9]
式中,*符号表示与铱键合的碳。
[化10]
式中,*符号表示与铱键合的碳。
[化11]
式中,*符号表示与铱键合的碳。
[化12]
式中,*符号表示与铱键合的碳。
[化13]
式中,*符号表示与铱键合的碳。
[化14]
式中,*符号表示与铱键合的碳。
[化15]
式中,*符号表示与铱键合的碳。
[化16]
式中,*符号表示与铱键合的碳。
以上所说明的本发明的有机铱配合物具有高发光效率,将其在高分子薄膜中掺入0.05mmol/g时的内部量子效率ΦPL为0.45以上。因此,适合作为发光层安装在有机电致发光元件中。
另外,对于本发明的有机铱配合物,其在发光色为黄色至绿色的范围内,在高分子薄膜中特别容易显示出高发光量子产率。具体而言,高分子薄膜中的发光波长(λPL)优选为510nm以上580nm以下,特别优选为510nm以上550nm以下。
对于本发明的有机铱配合物,可在通过使铱盐与构成C-N配位基的含氮化合物进行加热反应而得到前驱体之后,使该前驱体与β-二酮化合物进行加热反应来合成。此外,对于本发明的有机铱配合物,也可通过使金属盐与β-二酮化合物反应后,再使含氮化合物进行反应而合成。用来得到前驱体的加热反应优选在80℃以上130℃以下进行12小时以上24小时以下;与β-二酮的加热反应优选在60℃以上130℃以下进行0.5小时以上12小时以下。反应优选在溶剂存在下进行。上述合成反应所使用的铱盐优选为氯化物(IrCl3)。另外,作为其使用形态,可使用氯化物的水合物。
在以上所说明的有机铱配合物应用在有机EL元件的情况下,可通过旋转涂布法、真空沉积法等方法形成发光层。利用旋转涂布法来形成元件较简便且成本较低。
[发明的效果]
本发明的有机铱配合物在高分子薄膜中的发光量子产率高,适合用作有机EL元件的发光材料。
附图说明
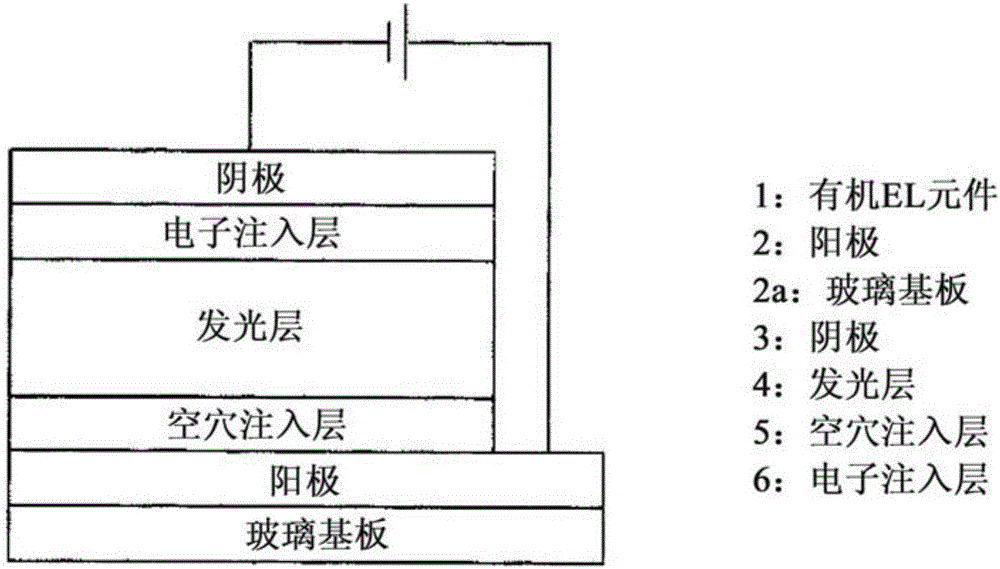
[图1]实施方案中的有机铱配合物的发光特性评价结果。
[图2]实施方案中所制作的有机EL元件的剖面示意图。
[图3]实施方案中的有机EL元件的发光特性评价结果。
具体实施方式
以下说明本发明的最佳实施方案。
合成以下所示的具有各配位基的有机铱配合物,并对所得的配合物进行发光光谱的测定及发光量子产率的评价。X为常规例的β-二酮配位基。可在使铱盐与含氮化合物进行反应而得到前驱体之后,使前驱体与β-二酮化合物进行反应,从而合成各铱化合物。
[化17]
首先,说明上述各铱配合物的合成方法概要。首先,合成各β-二酮化合物及各C-N配位基(1:2’,6’-二氟-2,3’-联吡啶)、(2:2-(3,5-双(三氟甲基)苯基)吡啶)、(3:2-(2,4-双(三氟甲基)苯基)吡啶)。接着,使各C-N配位基分别与氯化铱进行反应从而合成前驱体(1)、(2)及(3)。通过使各前驱体与β-二酮化合物进行反应,从而得到上述各铱配合物。
初始原料及合成所使用的试剂及溶剂均使用未进行精制的市售试剂等级。对于二苯甲酰甲烷(β-二酮(X)),也是直接将市售品用于配合物的合成。对于干燥四氢呋喃,购入市售的脱水四氢呋喃直接使用。另外,使用关东化学公司制的球状硅胶(中性),作为柱层析法所使用的填充剂。
使用质子核磁共振(1H NMR)光谱及质量分析(质(MS)谱)来鉴别所合成的配合物。使用Jeol JNM-ECX400分光光度计(400MHz)或Jeol JNM-ECS400分光光度计(400MHz)来测定1H NMR光谱。对于MS光谱,则以电喷雾电离法(ESI法)或基质辅助激光解吸离子化法(MALDI法)将试样离子化,并通过飞行时间(TOF)型质量分析计进行测定(ESI-TOF-MS及MALDI-TOF-MS光谱)。此外,在MALDI-TOF-MS光谱中,使用α-氰基-4-羟基肉桂酸(CHCA)作为基质。ESI-TOF-MS测定使用Jeol JML-T100LP质量分析装置,MALDI-TOF-MS测定使用Shimadzu-Kratos AXIMA-CFR PLUS TOF Mass质量分析装置。对于元素分析,以乙酰苯胺为标准物质,通过J-Science MICRO CORDER JM-10分析装置进行测定。
β-二酮化合物(A)的合成
由3,5-二(叔丁基)苯甲酸合成3,5-二(叔丁基)苯甲酸甲酯与3’,5’-二(叔丁基)苯乙酮后,通过使用该两个化合物的合成反应,得到β-二酮化合物(A)。
<3,5-二(叔丁基)苯甲酸甲酯的合成>
在氮气气氛下,在0℃下将浓硫酸(0.9mL)滴至3,5-二(叔丁基)苯甲酸(3.00g,12.8mmol)与甲醇(9mL)的混合物中,之后,一边搅拌一边使其加热回流1小时。冷却后,加入氯仿(100mL),再加入水(100mL)并在分液漏斗内进行震荡,从而分离有机层。重复进行一次该操作后,将分离的有机层合并为一。再以饱和碳酸氢钠水溶液(50mL)及饱和食盐水(50mL)清洗该有机层后,加入适量无水硫酸镁以使其干燥。通过过滤除去硫酸镁后,以蒸发器馏去溶剂,并在减压下使残渣在干燥器内进行干燥,由此得到3,5-二(叔丁基)苯甲酸甲酯。所得化合物为白色固体,其产率为92%(2.92g,11.8mmol)。以上述方法合成的化合物的特性(1H NMR、TOF MS)如下所示。
1H NMR(CDCl3):δ1.35(s,18H),3.91(s,3H),7.62(t,J=2.0Hz,1H),7.89(d,J=2.0Hz,2H)。
MALDI-TOF MS:m/z 249([M+H]+)。
[化18]
<3’,5’-二(叔丁基)苯乙酮的合成>
将3’,5’-二(叔丁基)苯甲酸(3.00g,12.8mmol)加入到干燥四氢呋喃(120mL)中,在氮气气氛下搅拌同时进行冷却,直到变成0℃以下为止。将甲基锂的3.0M的二乙氧基甲烷溶液(15mL)滴加至该混合物,之后升温至室温并搅拌2小时。将6M的盐酸加入到该反应混合物从而使其成为酸性后,以氯仿(100mL×2)进行萃取。使所得的有机层合并为一,并以水(50mL×2)、饱和碳酸氢钠水溶液(50mL)及饱和食盐水(50mL)进行清洗后,加入适量无水硫酸镁从而进行干燥。通过过滤除去硫酸镁后,以蒸发器馏去溶剂,以硅胶柱层析法(展开溶剂:氯仿)对残渣进行精制,由此得到3’,5’-二(叔丁基)苯乙酮。所得的化合物为无色液体,其产率为75%(2.23g,9.60mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3)d1.37(s,18H),2.60(s,3H),7.64(t,J=1.6Hz,1H),7.80(d,J=1.6Hz,2H)。
MALDI-TOF MS:m/z 232(M+)。
[化19]
<β-二酮化合物(A)的合成>
将3,5-二(叔丁基)苯甲酸甲酯(2.92g,11.8mmol)与氢化钠(60%油分散;1.27g,31.8mmol)加入到干燥THF(23mL)中,在氮气气氛下,在室温进行搅拌。在30分钟之内,向其中滴加使3’,5’-二(叔丁基)苯乙酮(2.23g,9.60mmol)溶解于干燥THF(23mL)所得的溶液。之后,在60℃下搅拌所得的反应混合物24小时。冷却后,加入1M的盐酸使其成为酸性后,以氯仿(100mL×2)进行萃取。将所得的有机层合并为一,并以水(50mL×2)、饱和碳酸氢钠水溶液(50mL)及饱和食盐水(50mL)进行清洗后,加入适量无水硫酸镁从而进行干燥。通过过滤除去硫酸镁后,以蒸发器馏去溶剂,以硅胶柱层析法(展开溶剂:氯仿)对残渣进行精制,由此得到1,3-双(3,5-二(叔丁基)苯基)丙烷-1,3-二酮(β-二酮A)。所得的化合物为琥珀色的浆状物质,其产率为49%(2.12g,4.73mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3)d1.38(s,36H),6.78(s,1H),7.63(t,J=2.0Hz,2H),7.78(d,J=2.0Hz,4H),16.9(brs,1H)。
MALDI-TOF MS:m/z 449([M+H]+)。
[化20]
β-二酮化合物(B)的合成
通过使用1,2,3,4-四氢-1,1,4,4-四甲基萘与丙二酰氯的合成反应,得到β-二酮化合物(B)。
<β-二酮化合物(B)的合成>
将1,2,3,4-四氢-1,1,4,4-四甲基萘(5.00g,26.6mmol)、丙二酰氯(1.35g,9.58mmol)及氯化铝(5.51g,41.3mmol)添加到二硫化碳(27mL)中,并在50℃下加热搅拌。接着,添加冷却的2mol/L的盐酸(27mL)并移至分液漏斗,以氯仿进行萃取。进一步以水清洗有机层,并以蒸发器馏去溶剂后,在残渣中加入浓盐酸(3.5mL)与氯仿(35mL),使其加热回流9小时。冷却后,将混合物移至分液漏斗,并以水及饱和食盐水进行清洗。以无水硫酸镁使有机层干燥后,以旋转蒸发器馏去溶剂。以硅胶柱层析法(展开溶剂;乙酸乙酯:己烷=1:2(v/v))对残渣进行精制,由此以39%的产率得到β-二酮(B)(1.66g,3.74mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.30(s,12H),1.34(s,12H),1.71(m,8H),6.76(s,1H),7.40(d,J=8.0Hz,2H),7.68(dd,J=8.0及2.0Hz,2H),7.94(d,J=2.0Hz,2H),16.96(brs,1H)。
MALDI-TOF MS:m/z 445([M+H]+)。
[化21]
接着,通过以下内容,合成C-N配位基(1)至(4)。
<C-N配位基(1)的合成>
依照下式,使2,6-二氟吡啶硼酸与2-溴基吡啶进行反应从而得到C-N配位基(1)。在氮气环境下,使由2,6-二氟吡啶硼酸(2.08g,13.1mmol)、2-溴基吡啶(1.34g,8.48mmol)、THF(65mL)、水(26mL)、K2CO3(1.51g,10.9mmol)及Pd(PPh3)4(0.67g,0.580mmol)所构成的混合物加热回流16小时。冷却后,以旋转蒸发器浓缩至液量约为三分之一,并将所得的混合物移至分液漏斗。以适量的氯仿稀释后,以水及饱和食盐水进行清洗,并以无水硫酸镁使有机层干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去溶剂。以硅胶柱层析法(展开溶剂;乙酸乙酯:己烷=1:3(v/v))对得到的残渣进行精制,由此以93%的产率得到C-N配位基(1)(1.51g,7.86mmol)。以上述方法合成的化合物的1H NMR特性如下所示。
1H NMR(CDCl3):δ6.93(dd,J=3.2及8.4Hz,1H),7.20-7.32(m,1H),7.68-7.86(m,2H),8.56-8.73(m,2H)。
[化22]
<C-N配位基(2)的合成>
依照下式,使3,5-双(三氟甲基)苯基硼酸与2-碘吡啶进行反应从而得到C-N配位基(2)。在氮气环境下,使由3,5-双(三氟甲基)苯基硼酸(2.00g,7.75mmol)、2-碘吡啶(1.17g,5.71mmol)、THF(30mL)、水(10mL)、K2CO3(4.50g,32.6mmol)及Pd(PPh3)4(0.261g,0.226mmol)所构成的混合物加热回流48小时。冷却后,以旋转蒸发器浓缩至液量约为三分之一,并将所得的混合物移至分液漏斗。以适量的氯仿稀释后,以水及饱和食盐水进行清洗,并以无水硫酸镁使有机层干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去溶剂。以硅胶柱层析法(展开溶剂;氯仿)对得到的残渣进行精制,由此以51%的产率得到C-N配位基(2)(0.850g,2.92mmol)。以上述方法合成的化合物的1H NMR特性如下所示。
1H NMR(CDCl3):δ7.34-7.38(m,1H),7.81-7.87(m,2H),7.92(s,1H),8.49(s,2H),8.76(dt,J=2.2及5.0Hz,1H)。
[化23]
<C-N配位基(3)的合成>
依照下式,使2,4-双(三氟甲基)苯基硼酸与2-碘吡啶进行反应从而得到C-N配位基(3)。在氮气环境下,使由2,4-双(三氟甲基)苯基硼酸(2.31g,8.96mmol)、2-碘吡啶(1.16g,5.66mmol)、苯(25mL)、乙醇(10mL)、K2CO3(7.31g,52.9mmol)及PdCl2(PPh3)2(0.383g,0.546mmol)所构成的混合物加热回流16小时。冷却后,将反应混合物移至分液漏斗,并以适量的氯仿进行稀释。以水及饱和食盐水清洗该混合液,并以无水硫酸镁使有机层干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去溶剂。以硅胶柱层析法(展开溶剂;氯仿)对得到的残渣进行精制,由此以59%的产率得到C-N配位基(3)(0.978g,3.36mmol)。以上述方法合成的化合物的1H NMR特性如下所示。
1H NMR(CDCl3):δ7.33(ddd,J=7.8,7.7及1.1Hz,1H),7.42(d,J=7.8Hz,1H),7.64(d,J=7.7Hz,1H),7.76(ddd,7.7,7.7及1.9Hz,1H),7.86(d,J=8.3Hz,1H),8.01(s,1H)。
[化24]
<C-N配位基(4)的合成>
依照下式,使2,4-二氟-3-氰基苯基硼酸与2-碘吡啶进行反应从而得到C-N配位基(4)。在氮气环境下,使由2,4-二氟吡啶硼酸(0.976g,5.34mmol)、2-碘吡啶(0.733g,3.58mmol)、苯(15mL)、乙醇(6mL)、水(15mL)、K2CO3(4.56g,33.0mmol)及Pd(PPh3)2Cl2(0.215g,0.306mmol)所构成的混合物加热回流18小时。冷却后,以旋转蒸发器浓缩至液量约为三分之一,并将所得的混合物移至分液漏斗。以适量的氯仿稀释后,以水及饱和食盐水进行清洗,并以无水硫酸镁使有机层干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去溶剂。以硅胶柱层析法(展开溶剂;氯仿)对得到的残渣进行精制,由此以80%的产率得到C-N配位基(4)(0.620g,2.87mmol)。以上述方法合成的化合物的1H NMR特性如下所示。
1H NMR(CDCl3):δ7.18(ddd,J=1.4,7.8及9.2Hz,1H),7.33(ddd,J=1.4,5.0及7.3Hz,1H),7.76-7.84(m,2H),7.86(td,J=6.4及8.7Hz,1H),8.72(m,1H)。
[化25]
使上述所合成的各C-N配位基与氯化铱进行反应,从而得到前驱体(1)至(3)。
<前驱体(1)的合成>
依照下式,使C-N配位基(1)与氯化铱进行反应从而得到前驱体(1)。将2’,6’-二氟-2,3’-联吡啶(0.500g,2.60mmol)、氯化铱三水合物(0.442g,1.25mmol)、水(17mL)及2-乙氧基乙醇(48mL)的混合物在100℃下搅拌12小时。冷却后,以旋转蒸发器进行浓缩,并向混合物中加入水(50mL)。进行抽吸过滤并回收生成的沉淀,以适量的甲醇进行清洗,由此以73%的产率得到前驱体(1)(0.580g,0.475mmol)。所得的化合物为难溶性固体。不进行进一步精制,而将其用于下述铱配合物的合成。
[化26]
<前驱体(2)的合成>
依照下式,使C-N配位基(2)与氯化铱进行反应从而得到前驱体(2)。将2-(3,5-双(三氟甲基)苯基)吡啶(0.850g,2.92mmol)、氯化铱三水合物(0.430g,1.22mmol)、水(20mL)及2-乙氧基乙醇(48mL)的混合物在100℃下搅拌12小时。冷却后,以旋转蒸发器进行浓缩,并向混合物中加入水(50mL)。进行抽吸过滤并回收生成的沉淀,以适量的甲醇进行清洗,由此以79%的产率得到前驱体(2)(0.930g,0.575mmol)。所得的化合物为难溶性固体。不进行进一步精制,而将其用于下述铱配合物的合成。
[化27]
<前驱体(3)的合成>
依照下式,使C-N配位基(3)与氯化铱进行反应从而得到前驱体(3)。将2-(2,4-双(三氟甲基)苯基)吡啶(0.431g,1.48mmol)、氯化铱三水合物(0.264g,0.749mmol)、水(10mL)及2-乙氧基乙醇(28mL)的混合物在100℃下搅拌12小时。冷却后,以旋转蒸发器进行浓缩,并向混合物中加入水(30mL)。进行抽吸过滤并回收生成的沉淀,以适量的甲醇进行清洗,由此以55%的产率得到前驱体(3)(0.329g,0.204mmol)。所得的化合物为难溶性固体。不进行进一步精制,而将其用于下述铱配合物的合成。
[化28]
<前驱体(4)的合成>
依照下式,使C-N配位基(4)与氯化铱进行反应从而得到前驱体(4)。将2,6-二氟-3-(吡啶-2-基)苯甲腈(0.612g,2.83mmol)、氯化铱三水合物(0.492g,1.40mmol)、水(21mL)及2-乙氧基乙醇(64mL)的混合物在100℃下搅拌25小时。冷却后,以旋转蒸发器进行浓缩,并向混合物中加入水(50mL)。进行抽吸过滤并回收生成的沉淀,以适量的甲醇进行清洗,由此以83%的产率得到前驱体(4)(0.762g,0.579mmol)。所得的化合物为难溶性固体。不进行进一步精制,而将其用于下述铱配合物的合成。
[化29]
接着,使以上所合成的前驱体(1)至(4)与β-二酮(A)、(B)及(X)分别进行反应,得到各铱配合物。
铱配合物(1-A)的合成
依照下式,使前驱体(1)与β-二酮(A)进行反应从而得到铱配合物1-A。
[化30]
<铱配合物(1-A)的合成>
将前驱体(1)(0.244g,0.200mmol)、1,3-双(3,5-二(叔丁基)苯基)丙烷-1,3-二酮(β-二酮(A))(0.131g,0.292mmol)及碳酸钠(1.90g,17.9mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在80℃下搅拌该混合物30分钟。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;二氯甲烷)对所得的残渣进行精制,更进一步以乙腈进行重结晶,由此以25%的产率得到铱配合物1-A(73.5mg,0.0719mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.26(s,36H),5.81(t,J=1.8Hz,2H),6.58(s,1H),7.19-7.22(m,2H),7.53(m,6H),7.85-7.89(m,2H),8.29(d,J=7.6Hz,2H),8.57(dd,J=0.80及6.0Hz,2H)。
ESI-TOF MS:m/z 1045([M+Na]+)。
C51H53F4IrN4O2分析计算值:C,59.92;H,5.23;5.48。实际测量值:C,60.10;H,5.03;5.50。
铱配合物(1-B)的合成
依照下式,使前驱体(1)与β-二酮(B)进行反应从而得到铱配合物1-B。
[化31]
<铱配合物(1-B)的合成>
将前驱体(1)(0.246g,0.201mmol)、β-二酮(B)(0.129g,0.290mmol)及碳酸钠(1.90g,17.9mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在80℃下搅拌该混合物30分钟。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;二氯甲烷)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以25%的产率得到铱配合物1-B(75.1mg,0.0738mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.23(s,12H),1.25(s,12H),1.66(s,8H),5.76(s,2H),6.58(s,1H),7.17-7.21(m,2H),7.29(d,J=8.0Hz,2H),7.49-7.52(m,2H),7.71(d,J=2.0Hz,2H),7.83-7.87(m,2H),8.27(d,J=8.8Hz,2H),8.53(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 1041([M+Na]+)。
C51H49F4IrN4O2分析计算值:C,60.16;H,4.85;5.50。实际测量值:C,60.12;H,4.61;5.45。
铱配合物(1-X)的合成
依照下式,使前驱体(1)与β-二酮(X)进行反应从而得到铱配合物1-X。
[化32]
<铱配合物(1-X)的合成>
将前驱体(1)(0.500g,0.410mmol)、β-二酮(X)(0.435g,1.94mmol)及碳酸钠(0.389g,3.67mmol)添加到2-乙氧基乙醇(77mL)中,在氮气气氛下,在105℃下搅拌该混合物2小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;二氯甲烷)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以38%的产率得到铱配合物1-X(250mg,0.313mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ5.75(s,2H),6.66(s,1H),7.23-7.25(m,2H),7.35(t,J=7.8Hz,4H),7.45-7.47(m,2H),7.77(dd,J=1.2及7.8Hz,4H),7.88(t,J=7.8Hz,2H),8.29(d,J=8.0Hz,2H),8.52(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 821([M+Na]+)。
C35H21F4IrN4O2分析计算值:C,52.69;H,2.65;7.02。实际测量值:C,52.92;H,2.73;7.01。
铱配合物(2-A)的合成
依照下式,使前驱体(2)与β-二酮(A)进行反应从而得到铱配合物2-A。
[化33]
<铱配合物(2-A)的合成>
将前驱体(2)(0.245g,0.152mmol)、1,3-双(3,5-二(叔丁基)苯基)丙烷-1,3-二酮(β-二酮(A))(0.129g,0.288mmol)及碳酸钠(1.90g,17.9mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在100℃下搅拌该混合物2小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿:己烷=1:1(v/v))对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以5.8%的产率得到铱配合物2-A(20.3mg,0.0166mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.24(s,36H),6.34(s,1H),6.94-6.97(m,2H),7.30(d,J=1.6Hz,4H),7.48(t,J=1.6Hz,2H),7.55(brs,2H),7.78(dt,J=1.6及7.8Hz,2H),8.05(d,J=7.6Hz,2H),8.16(brs,2H),8.27(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 1243([M+Na]+)。
C57H55F12IrN2O2分析计算值:C,56.10;H,4.54;2.30。实际测量值:C,55.93;H,4.49;2.26。
铱配合物(2-B)的合成
依照下式,使前驱体(2)与β-二酮(B)进行反应从而得到铱配合物2-B。
[化34]
<铱配合物(2-B)的合成>
将前驱体(2)(0.154g,0.0953mmol)、β-二酮(B)(0.200g,0.450mmol)及碳酸钠(0.0900g,0.849mmol)添加到2-乙氧基乙醇(18mL)中,在氮气气氛下,在105℃下搅拌该混合物3小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以39%的产率得到铱配合物2-B(90.0mg,0.0740mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.16(s,12H),1.23(s,12H),1.64(s,8H),6.32(s,1H),6.91-6.95(m,2H),7.22(d,J=7.8Hz,2H),7.30(dd,J=2.0及7.8Hz,2H),7.43(d,J=2.0Hz,2H),7.55(s,2H),7.73-7.78(m,2H),8.01(d,J=8.0Hz,2H),8.16(s,2H),8.22(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 1239([M+Na]+)。
C57H51F12IrN2O2分析计算值:C,56.29;H,4.23;2.30。实际测量值:C,56.60;H,4.40;2.26。
铱配合物(2-X)的合成
依照下式,使前驱体(2)与β-二酮(X)进行反应从而得到铱配合物2-X。
[化35]
<铱配合物(2-X)的合成>
将前驱体(2)(0.331g,0.205mmol)、β-二酮(X)(0.220g,0.981mmol)及碳酸钠(0.195g,1.84mmol)添加到2-乙氧基乙醇(39mL)中,在氮气气氛下,在105℃下搅拌该混合物3小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以30%的产率得到铱配合物2-X(120mg,0.121mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ6.43(s,1H),6.94-6.98(m,2H),7.29(t,J=7.8Hz,4H),7.41(t,J=7.8Hz,2H),7.55-7.57(m,6H),7.74-7.78(m,2H),8.00(d,J=8.4Hz,2H),8.13(s,2H),8.21(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 1019([M+Na]+)。
C41H23F12IrN2O2·H2O分析计算值:C,48.57;H,2.49;2.76。实际测量值:C,48.76;H,2.60;2.67。
铱配合物(3-A)的合成
依照下式,使前驱体(3)与β-二酮(A)进行反应从而得到铱配合物3-A。
[化36]
<铱配合物(3-A)的合成>
将前驱体(3)(0.240g,0.149mmol)、1,3-双(3,5-二(叔丁基)苯基)丙烷-1,3-二酮(β-二酮(A))(0.129g,0.288mmol)及碳酸钠(1.91g,18.1mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在100℃下搅拌该混合物2小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿:己烷=1:1(v/v))对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以44%的产率得到铱配合物3-A(154mg,0.126mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.25(s,36H),6.53(s,1H),6.70(s,2H),7.20-7.24(m,2H),7.44(d,J=2.0Hz,4H),7.50(t,J=2.0Hz,2H),7.54(s,2H),7.86-7.91(m,2H),8.44(d,J=8.0Hz,2H),8.67(dd,J=1.4及5.8Hz,2H)。
ESI-TOF MS:m/z 1243([M+Na]+)。
C57H55F12IrN2O2分析计算值:C,56.10;H,4.54;2.30。实际测量值:C,56.27;H,4.74;2.41。
铱配合物(3-B)的合成
依照下式,使前驱体(3)与β-二酮(B)进行反应从而得到铱配合物3-B。
[化37]
<铱配合物(3-B)的合成>
将前驱体(3)(0.241g,0.149mmol)、β-二酮(B)(0.129g,0.290mmol)及碳酸钠(1.90g,18.0mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在100℃下搅拌该混合物2小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿:己烷=1:1(v/v))对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以17%的产率得到铱配合物2-B(59.7mg,0.0491mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.16(s,6H),1.18(s,6H),1.22(s,6H),1.23(s,6H),1.64(s,8H),6.52(s,1H),6.64(s,2H),7.20(t,J=6.0Hz,2H),7.27(d,J=7.8Hz,2H),7.43(dd,J=2.0及7.8Hz,2H),7.53(s,2H),7.61(d,J=2.0Hz,2H),7.83-7.88(m,2H),8.41(d,J=8.0Hz,2H),8.63(d,J=6.0Hz,2H)。
ESI-TOF MS:m/z 1239([M+Na]+)。
C57H51F12IrN2O2分析计算值:C,56.29;H,4.23;2.30。实际测量值:C,56.30;H,4.05;2.28。
铱配合物(3-X)的合成
依照下式,使前驱体(3)与β-二酮(X)进行反应从而得到铱配合物3-X。
[化38]
<铱配合物(3-X)的合成>
将前驱体(3)(0.200g,0.124mmol)、β-二酮(X)(0.128g,0.571mmol)及碳酸钠(0.115g,1.09mmol)添加到2-乙氧基乙醇(23mL)中,在氮气气氛下,在105℃下搅拌该混合物3小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入氯仿。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸钠从而使其干燥。通过过滤除去硫酸钠后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以10%的产率得到铱配合物3-X(25.0mg,0.0251mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ6.60(s,2H),6.61(s,1H),7.21-7.24(m,2H),7.32(t,J=7.4Hz,4H),7.44(t,J=7.4Hz,2H),7.54(s,2H),7.72(d,J=7.4Hz,4H),7.85-7.90(m,2H),8.41(d,J=8.8Hz,2H),8.64(dd,J=0.8及5.8Hz,2H)。
ESI-TOF MS:m/z 1019([M+Na]+)。
C41H23F12IrN2O2分析计算值:C,49.45;H,2.33;2.81。实际测量值:C,49.60;H,2.70;2.70。
铱配合物(4-A)的合成
依照下式,使前驱体(4)与β-二酮(A)进行反应从而得到铱配合物4-A。
[化39]
<铱配合物(4-A)的合成>
将前驱体(4)(0.243g,0.185mmol)、1,3-双(3,5-二(叔丁基)苯基)丙烷-1,3-二酮(β-二酮(A))(0.133g,0.296mmol)及碳酸钠(1.90g,17.9mmol)添加到2-乙氧基乙醇(50mL)中,在氮气气氛下,在100℃下搅拌该混合物2小时。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入乙酸乙酯。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸镁从而使其干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;氯仿)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以12%的产率得到铱配合物4-A(00.459g,0.0429mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.26(s,36H),5.98(d,J=8.6Hz,2H),6.56(s,1H),7.22(td,J=1.4及5.9Hz,2H),7.49(d,J=1.8Hz,4H),7.54(t,J=1.8Hz,2H),7.90(td,J=1.4及8.2Hz,2H),8.32(d,J=8.2Hz,2H),8.55(dd,J=1.4及5.9Hz,2H)。
ESI-TOF MS:m/z 1093([M+Na]+)。
C55H53F4IrN4O2分析计算值:C,61.72;H,4.99;N,5.23。实际测量值:C,61.72;H,5.03;N,5.23。
铱配合物(4-B)的合成
依照下式,使前驱体(4)与β-二酮(B)进行反应从而得到铱配合物4-B。
[化40]
<铱配合物(4-B)的合成>
将前驱体(4)(0.106g,0.0805mmol)、β-二酮(B)(0.0884g,0.199mmol)及碳酸钠(0.0531g,0.501mmol)添加到2-乙氧基乙醇(25mL)中,在氮气气氛下,在40℃下搅拌该混合物1小时30分钟。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入二氯甲烷。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸镁从而使其干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;二氯甲烷)对所得的残渣进行精制,进一步以甲醇进行重结晶,由此以22%的产率得到铱配合物4-B(0.0378g,0.0355mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.24(m,24H),1.66(m,8H),5.92(d,J=8.6Hz,2H),6.58(s,1H),7.21-7.24(m,2H),7.29(d,J=8.6Hz,2H),7.48(dd,J=1.8及8.6Hz,2H),7.69(d,J=1.8Hz,2H),7.86-7.90(m,2H),8.30(d,J=8.6Hz,2H),8.50(dd,J=1.4及5.9Hz,2H)。
ESI-TOF MS:m/z 1089([M+Na]+)。
C55H49F4IrN4O2分析计算值:C,61.96;H,4.63;N,5.25。实际测量值:C,61.94;H,4.59;N,5.25。
铱配合物(4-X)的合成
依照下式,使前驱体(4)与β-二酮(X)进行反应从而得到铱配合物4-X。
[化41]
<铱配合物(4-X)的合成>
将前驱体(4)(0.0980g,0.0745mmol)、β-二酮(X)(0.0496g,0.221mmol)及碳酸钠(0.0650g,0.613mmol)添加到2-乙氧基乙醇(7mL)中,在氮气气氛下,在40℃下搅拌该混合物30分钟。冷却后,以旋转蒸发器馏去溶剂,并向残渣中加入二氯甲烷。以水和饱和食盐水清洗所得的混合溶液,并加入适量无水硫酸镁从而使其干燥。通过过滤除去硫酸镁后,以旋转蒸发器馏去滤液的溶剂。以硅胶柱层析法(展开溶剂;二氯甲烷)对所得的残渣进行精制,更进一步以甲醇进行重结晶,由此以21%的产率得到铱配合物4-X(0.0254g,0.0300mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ5.91(d,J=8.8Hz,2H),6.65(s,1H),7.26-7.28(m,2H),7.35(t,J=7.7Hz,4H),7.45-7.49(m,2H),7.75-7.77(m,4H),7.90(td,J=1.4及8.4Hz,2H),8.32(d,J=8.4Hz,2H),8.49(dd,J=1.4及5.6Hz,2H)。
ESI-TOF MS:m/z 869([M+Na]+)。
C39H21F4IrN4O2分析计算值:C,55.38;H,2.50;N,6.62。实际测量值:C,55.38;H,2.74;N,6.74。
[发光(PL)光谱及PL量子产率的评价]
使用上述所得的各铱配合物,测定其发光(PL)光谱及PL量子产率ΦPL。使用“堀場製作所社”制的Fluorolog-3分光光度计来进行PL光谱的测定。使用“浜松ホトニクス社”制的C9920-12量子产率测定装置来进行PL量子产率的测定。对于这些PL光谱及PL量子产率的评价,在作为介质的高分子薄膜(聚甲基丙烯酸甲酯,PMMA)中进行评价。需要说明的是,关于溶液试样,封入氩气作为脱氧溶液进行测定,关于高分子薄膜试样,在氮气气氛下进行测定。关于高分子薄膜试样,在PMMA中掺入0.05mmol/g(4重量%)的各铱配合物从而进行测定。结果显示于下表。
[表1]
由上表可知,具有经叔丁基取代的苯基作为β-二酮的铱配合物(1-A、2-A、3-A、4-A、1-B、2-B、3-B、4-B)在高分子薄膜中显示出超过0.45的高发光量子产率。
接着,对于铱配合物1-A、2-A、1-B、2-B,以有机溶剂(二氯甲烷(CH2Cl2))作为介质进行PL光谱及PL量子产率的评价。结果示于下表及图1。下表中一起显示了高分子薄膜中的结果,同时显示有机溶剂中的波长峰值位置与高分子薄膜中的波长峰值位置的差(波长位移ΔλPL)的计算值。
[表2]
由上表及图1可知,对于具有经叔丁基取代的苯基作为β-二酮的铱配合物(1-A、2-A、1-B、2-B),相对于有机溶剂中的波长,可发现在高分子薄膜中峰值位置位移至短波长侧的刚致变色。作为发光色,在有机溶剂中为黄绿色至橙色的发光波长,但在高分子薄膜中变成绿色至黄色的发光波长。
由以上结果可知,对于具有经叔丁基取代的苯基作为β-二酮的铱配合物,将其在高分子薄膜中掺入0.05mmol时的PL量子产率ΦPL为0.45以上,同时相对于有机溶剂中的发光波长,显示出在高分子薄膜中发光波长位移至短波长侧的刚致变色。
[有机EL元件的制作与特性评价]
使用上述各铱配合物,以下述顺序制作图2所示的有机EL元件(1),并进行特性评价。
<有机EL元件的制作>
(a)空穴注入层(5)的形成
对ITO-玻璃基板(“三容真空工業”制,ITO,膜厚150nm)实施图案化处理,并进行清洗,由此制备阳极(2)。接着,以臭氧对ITO薄膜进行表面处理。表面处理后,通过旋转涂布法迅速地使空穴注入材料成膜在ITO膜上,在120℃下烧制1小时,由此形成厚度为40nm的空穴注入层(5)。使用包含PEDOT与PSS的导电性聚合物(Heraeus Clevios制,P VP CH8000)作为空穴注入材料。
(b)发光层(4)的形成
使聚(9-乙烯基咔唑)(PVCz,Sigma-Aldrich制,数均分子量Mn为25000至50000,通过从THF-甲醇再沉淀进行精制)、2-(4-联苯基)-5-(4-叔丁基苯基)-1,3,4-噁二唑(PBD)及铱配合物1-A溶解于脱水甲苯中,并以膜过滤器(“メルクミリポア”制,0.2μm Millex-FG)进行过滤,由此调制发光层用墨液Ink(1-A)。将PBD、铱配合物掺入到PVCz,并且使PBD和铱配合物的浓度相对于1g的PVCz分别为850μmol、98μmol,并相对于10mg的PVCz,使用0.7ml的甲苯作为墨液溶剂。使用所得的发光层用墨液Ink(1-A),通过旋转涂布法,使其成膜于空穴注入层(5)上,并在120℃烧制1小时,由此形成厚度为80nm的发光层4。
(c)电子注入层(6)及阴极(3)的形成
使用阴影掩模,通过真空沉积形成作为电子注入材料的氟化铯薄膜(电子注入层(6),厚度为1nm),接着,制作铝的薄膜(阴极(3),厚度为250nm)。此时,以使发光部的面积为10mm2(2mm×5mm)的方式来制作电子注入层(6)及阴极(3)。如此地,完成有机EL元件EL(1-A)。
<将各铱配合物用于发光材料的有机EL元件的制作>
使用铱配合物2-A代替铱配合物1-A来调制发光层用墨液Ink(2-A)。除了使用该发光层用墨液Ink(2-A)以外,以与上述相同的顺序,得到有机EL元件EL(2-A)。关于EL元件EL(3-A)、EL(4-A)、EL(1-B)、EL(2-B)、EL(3-B)、EL(4-B)、EL(1-X)、EL(2-X)、EL(3-X)、EL(4-X),也相同地使用配合物3-A、4-A、1-B、2-B、3-B、4-B、1-X、2-X、3-X、4-X来代替配合物1-A进行制作。
<有机EL元件特性的评价>
使用紫外线硬化树脂,将以上述步骤所得的有机EL元件密封于中空玻璃中,从而制作有机EL特性评价用的样品。
通过亮度光分布特性测定装置(“浜松ホトニクス社”制、C-9920-11)来测定EL光谱、最大亮度Lmax(cd/m2)、最大外部量子效率ηext,max(%)及CIE表色系统(x,y)等有机EL元件特性。
表2显示出有机EL元件的EL光谱的峰值波长λEL(nm)、最大亮度Lmax(cd/m2)、最大外部量子效率ηext,max(%)、最大电流效率ηj,max(cd/A)、最大功率效率ηp,max(lm/W)及CIE表色系统(x,y)的结果。关于Lmax及ηext,max,在()内也一并显示测定时的施加电压(V)。另外,发光开始电压Vturn-on表示亮度达到1cd/m2的电压。
另外,图3为有机EL元件EL(1-A)、EL(2-A)、EL(3-A)、EL(4-A)、EL(1-B)、EL(2-B)、EL(3-B)、EL(4-B)、EL(1-X)、EL(2-X)、EL(3-X)、EL(4-X)的电致发光(EL)光谱。EL光谱在最大亮度Lmax下进行测定。
[表3]
由上述结果可知,相较于使用配合物1-X、2-X、3-X、4-X所制作的有机EL元件,使用配合物1-A、2-A、3-A、4-A、1-B、2-B、3-B、4-B所制作的有机EL元件显示出良好的有机EL特性。
[工业实用性]
本发明的有机铱配合物在高分子薄膜中的发光量子产率高,适合作为有机EL元件的发光材料。本发明特别适合作为发出黄光至绿光的材料。
- 还没有人留言评论。精彩留言会获得点赞!