杂交探针和方法与流程
发明领域
本发明涉及用于检测、分析和处理核酸的组合物和方法。具体而言,本发明涉及用于产生和使用杂交探针的组合物和方法。
背景
fish(荧光原位杂交)是用于检测和定位特定dna序列在染色体上存在与否的细胞遗传学技术。fish使用这样的荧光探针,所述荧光探针仅结合它们与其显示高度序列互补性的染色体的那些部分。荧光显微术可用于发现荧光探针结合染色体的部位。fish经常用于发现用于遗传咨询、医学和物种鉴定的dna中的特定特征。fish也可以用于检测和定位细胞、循环肿瘤细胞和组织样品中的特定rna靶标(mrna、lncrna和mirna)。在本上下文中,其可以帮助确定在细胞和组织中基因表达的时空模式。
人基因组dna是在整个基因组中以多个拷贝存在的唯一序列和重复序列的混合物。在一些应用中,期望产生仅与染色体上的目标唯一序列退火的杂交探针。唯一序列探针的制备受到整个生物基因组中大量类别的重复序列的存在的扰乱(hood等人,molecμlarbiologyofeucaryoticcells(benjamin/cummingspublishingcompany,menlopark,calif.1975)。杂交探针中重复序列的存在降低了探针的特异性,因为探针的部分结合目标序列之外发现的其他重复序列。因此,为了确保杂交探针与特定目标序列的结合,需要缺少重复序列的探针。
近期的贡献通过使用未标记的阻断核酸抑制重复序列的杂交而解决这一问题(美国专利号5,447,841和美国专利号6,596,479)。在杂交中使用阻断核酸是昂贵的,不会完全阻止重复序列的杂交,并且可能使基因组杂交模式产生偏差(newkirk等人,“distortionofquantitativegenomicandexpressionhybridizationbycot-1dna:mitigationofthiseffect,”nucleicacidsres.vol33(22):el91(2005))。因此,需要不使用阻断dna而阻止重复序列的杂交的方法用于最优杂交。
实现这一点的一个方法是在杂交之前从杂交探针中去除不想要的重复区段。之前已经描述了涉及去除高度重复序列的技术。吸收剂如羟磷灰石提供用于从提取的dna中去除高度重复序列的方法。羟磷灰石层析基于双链体再结合条件诸如温度、盐浓度或其他严格性来将dna分级。这一程序是麻烦的且随不同序列而不同。重复dna也可以通过杂交至固定dna来去除(brison等人,“generalmethodsforcloningamplifieddnabydifferentialscreeningwithgenomicprobes,”molecularandcellularbiology,vol.2,pp.578-587(1982))。在所有这些程序中,物理去除重复序列将取决于基于dna序列的碱基组成用内在不同严格优化条件。
已经描述了用于从杂交探针中去除重复序列的若干其他方法。一种方法涉及使用交联剂来交联重复序列以直接阻止重复序列的杂交或以阻止重复序列在pcr反应中的扩增。(美国专利号6,406,850)。另一种方法使用pcr辅助的亲和层析以从杂交探针中去除重复(美国专利号6,569,621)。这两种方法都依赖于使用标记的dna来去除重复序列,这使得这些方法复杂且难以重现。进一步,两种方法都是耗时的,需要多轮重复去除以产生适合用于荧光原位杂交(fish)或其他需要高度靶特异性的杂交反应的功能探针。
因此,需要从探针中去除重复序列的方法。
发明概述
本发明涉及用于检测、分析和处理核酸的组合物和方法。具体而言,本发明涉及用于产生和使用杂交探针的组合物和方法。
本技术的实施方案提供用于产生和使用选择性产生或合成以排除非目标序列和/或包括目标序列的探针(例如,实质上不含重复片段的核酸探针)的组合物、试剂盒和系统。例如,在一些实施方案中,本发明提供产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp(例如,至少100、200、至少300或至少400)的目标核酸的不想要的序列(例如,无重复片段、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构或编码序列)且任选地彼此长度不超过20%不同(例如,20%或更少、10%或更少、5%或更少、4%或更少、3%或更少、3%或更少、1%或更少、或相同长度)的区域;和b)产生(例如经扩增、克隆、合成或其组合)对应于实质上不含不想要的序列的区域的多个含探针的核酸。在一些实施方案中,所述方法进一步包含以下步骤的一个或多个:c)将所述含探针的核酸片段化以产生探针;和d)进一步扩增探针的子集以产生实质上不含不想要的序列的探针(例如,缺少例如不想要的重复序列的ish探针)。在一些实施方案中,所述方法进一步包括d)根据大小分开探针的步骤。在一些实施方案中,所述分开使用层析或电泳进行。在一些实施方案中,所述方法进一步包括分离探针的子集的步骤。在一些实施方案中,所述子集基于分开的核酸的大小。在一些实施方案中,探针连接核酸衔接子。在一些实施方案中,衔接子是扩增引物。在一些实施方案中,将扩增引物功能化用于下游应用(例如通过添加标记、结合位点或限制位点)。在一些实施方案中,将探针分开和将探针子集分离。在一些实施方式中,扩增是pcr。在一些实施方案中,使用计算机软件和计算机处理器鉴定实质上不含不想要的序列的区域。在一些实施方案中,通过超声处理将含探针的核酸片段化(尽管可以使用多种其他化学、物理方法或其他方法的任一种)。在一些实施方案中,所述分开通过电泳或层析进行。在一些实施方案中,片段长度为约100至500bp,尽管也可以使用其他长度。在一些实施方案中,探针是标记的(例如,用荧光标记)。在一些实施方案中,探针为85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含不想要的核酸序列。
在一些实施方案中,本发明提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)将衔接子连接至探针;和任选地e)进一步扩增探针的子集。
进一步的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;和任选地d)进一步扩增探针的子集。
另外的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含不想要的序列的区域,其中所述不想要的区域为例如长度为至少100bp的重复序列、非保守序列、保守序列、富含gc的序列、富含at的序列、二级序列或编码序列;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;和c)将所述含探针的核酸片段化以产生探针;和任选地d)进一步扩增探针的子集。
仍其他的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)通过大小分开探针;e)分离探针的子集;和任选地e)进一步扩增探针的子集。
还其他的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)通过大小分开探针;e)分离探针的子集,其中所述子集包含长度为80-300bp的核酸(例如,长度大约150bp);和任选地e)进一步扩增探针的子集。
另外的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;任选地d)进一步扩增探针的子集产生探针组;和e)用所述探针组进行杂交测定(例如ish测定)。
本文进一步提供通过上述方法产生的不含不想要的序列的一组核酸探针(例如杂交探针(例如,原位杂交(ish)探针))和包含所述探针的试剂盒和系统。本公开不限于具体测定或靶标。在一些实施方案中,探针检测癌基因的表达或染色体非整倍性。
本文另外提供进行杂交测定的方法,包括将靶核酸与通过上述方法产生的探针(例如ish探针)接触。
本文还提供任何通过上述方法产生的探针(例如ish探针)在杂交(例如ish)测定中的用途。
本文描述了另外的实施方案。
附图说明
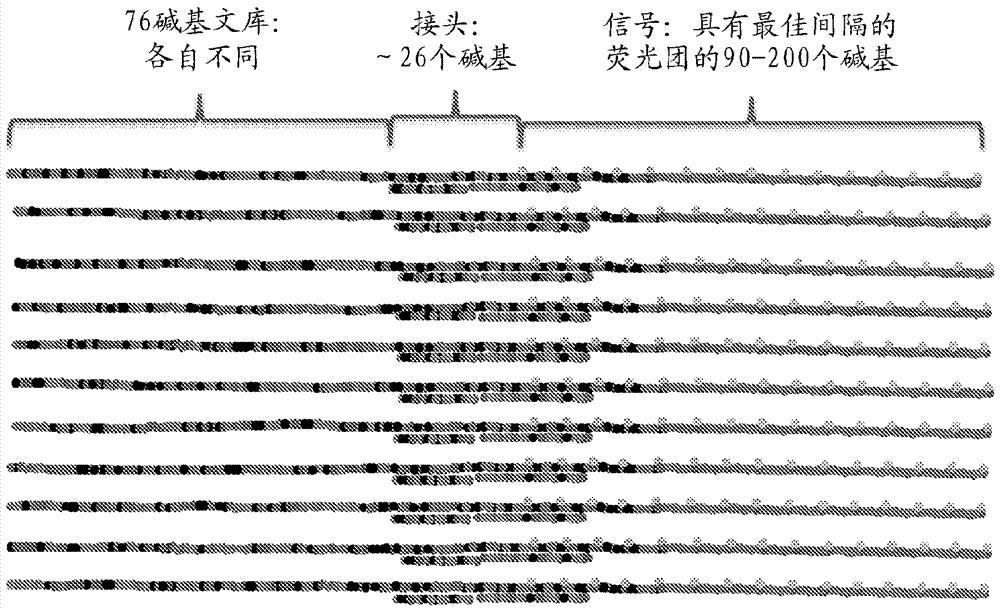
图1显示本发明的实施方案的示例性标记的探针的图示。
图2显示her3基因座的图谱。
图3显示从her基因座扩增无重复片段的探针。
图4显示来自her2基因座的片段化的探针。
图5显示衔接的(adapted)her2探针的扩增。
图6显示her2无重复片段探针。
图7显示使用本发明的实施方案的探针的her2基因座的fish杂交。
图8显示选择用于设计her-2探针的her-2基因的80bp部分。
图9显示示例性的无重复片段的p16探针的图谱。
定义
如本文所用,术语“实质上不含不想要的核酸”指实质上不含(例如85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含)不想要的核酸的核酸。不想要的核酸包括但不限于重复核酸、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构或编码序列。
如所用,术语“实质上不含重复片段(repeatfree)的核酸序列”或“不含重复片段(freeofrepeats)的核酸”指实质上不含(例如85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含)重复的核酸序列的核酸区域。
如本文所用,术语“样品”以其最广泛的含义使用。在一种含义中,其意指包括细胞(例如,人、细菌、酵母和真菌)、生物、获自任何来源的样本或培养物、以及生物样品。生物样品可以获自动物(包括人)并且指其中发现的生物材料或组合物,包括但不限于骨髓、血液、血清、血小板、血浆、间隙液、尿液、脑脊液、核酸、dna、组织、及其纯化或过滤形式。然而,这些实例不应解释为限定可用于本发明的样品类型。
如本文所用的术语“标记”指可用于提供可检测的(优选可定量的)效果且可以连接至核酸或蛋白的任何原子或分子。标记包括但不限于染料;放射性标记,诸如32p;结合部分诸如生物素;半抗原诸如地高辛;发光、发磷光或发荧光部分;和单独的荧光染料或与可以通过荧光共振能量转移(fret)抑制或移动发射光谱的部分组合的荧光染料。标记可以提供可通过荧光、放射性、比色、重量测定、x射线衍射或吸收、磁性、酶活性等检测的信号。标记可以是带电荷的部分(正电荷或负电荷)或可选地,可以是电荷中性的。标记可以包括核酸或蛋白序列或由其组合,只要包含标记的序列是可检测的。在一些实施方案中,核酸在没有标记的情况下直接检测(例如,直接读取序列)。
如文本所用,术语“核酸分子”指任何包含核酸的分子,包括但不限于dna或rna。该术语涵盖包含dna和rna的任何已知碱基类似物的序列,所述类似物包括但不限于:4-乙酰胞嘧啶、8-羟基-n6-甲基腺苷、吖丙啶基胞嘧啶、假异胞嘧啶、5-(羧基羟基甲基)尿嘧啶、5-氟尿嘧啶、5-溴尿嘧啶、5-羧甲基氨基甲基-2-硫代尿嘧啶、5-羧甲基氨基甲基尿嘧啶、二氢尿嘧啶、肌苷、n6-异戊烯基腺嘌呤、1-甲基腺嘌呤、1-甲基假尿嘧啶、1-甲基鸟嘌呤、1-甲基肌苷、2,2-二甲基鸟嘌呤、2-甲基腺嘌呤、2-甲基鸟嘌呤、3-甲基胞嘧啶、5-甲基胞嘧啶、n6-甲基腺嘌呤、7-甲基鸟嘌呤、5-甲基氨基甲基尿嘧啶、5-甲氧基氨基甲基-2-硫代尿嘧啶、β-d-甘露糖基q核苷、5'-甲氧基羰基甲基尿嘧啶、5-甲氧基尿嘧啶、2-甲硫基-n6-异戊烯基腺嘌呤、尿嘧啶-5-氧基乙酸甲酯、尿嘧啶-5-氧基乙酸、氧基丁氧基胸苷(oxybutoxosine)、假尿嘧啶、q核苷、2-硫胞嘧啶、5-甲基-2-硫尿嘧啶、2-硫尿嘧啶、4-硫尿嘧啶、5-甲基尿嘧啶、n-尿嘧啶-5-氧基乙酸甲酯、尿嘧啶-5-氧基乙酸、假尿嘧啶、q核苷、2-硫胞嘧啶和2,6-二氨基嘌呤。
如本文所用,术语“杂交”用于指互补核酸的配对。杂交和杂交强度(即核酸之间结合的强度)受此类因素如核酸之间的互补性程度、所涉及的条件的严格性、形成的杂交物的tm以及核酸内的g:c比率的影响。在其结构内含有互补核酸的配对的单一分子将是“自杂交的”。
术语“引物”指寡核苷酸,无论其是在经纯化的限制性消化物中天然存在或是合成产生的,所述寡核苷酸当置于诱导与核酸链互补的引物延伸产物合成的条件下时(例如,存在核苷酸和诱导剂诸如dna聚合酶和在合适的温度和ph下),能够作为合成的起始点而起作用。引物优选是单链的用于扩增的最大效率,但可选地也可以是双链的。如果是双链的,则在用于制备延伸产物前首先将引物处理以分开其链。优选地,引物是寡脱氧核糖核苷酸。引物应足够长以在诱导剂存在的情况下引发延伸产物的合成。引物的精确长度将取决于许多因素包括温度、引物来源和方法的使用。例如,在一些实施方案中,引物范围为10-100或更多个核苷酸(例如10-300、15-250、15-200、15-150、15-100、15-90、20-80、20-70、20-60、20-50个核苷酸等)。
在一些实施方案中,引物包含不与目标核酸杂交的另外的序列。术语“引物”包括化学修饰的引物、荧光修饰的引物、功能引物(融合引物)、序列特异性引物、随机引物、具有特异性和随机序列的引物、和dna和rna引物。
术语“扩增(amplifying或amplification)”在核酸的上下文中指产生多个拷贝的多核苷酸、或多核苷酸的部分,通常从少量的多核苷酸开始(例如,少至单个多核苷酸分子),其中扩增产物或扩增子通常是可检测的。多核苷酸的扩增包括多种化学和酶促方法。在聚合酶链反应(pcr)、滚环扩增(rca)或连接酶链反应(lcr)过程中从一个或几个拷贝的靶dna或模板dna分子产生多个dna拷贝是扩增的形式。扩增不限于起始分子的严格复制。例如,使用逆转录(rt)-pcr从样品中有限量的rna产生多个cdna分子是扩增的形式。此外,在转录过程期间从单一dna分子产生多个rna分子也是扩增的形式。
如本文所用,术语“固体支持物”用于指试剂诸如抗体、抗原和其他测试组分与其连接的任何固体或固定的材料。固体支持物的实例包括显微镜载玻片、微量滴定板的孔、盖玻片、珠粒、颗粒、细胞培养瓶以及任何其他合适的物品。
本发明实施方案详述
本发明涉及用于检测、分析和处理核酸的组合物和方法。具体而言,本发明涉及用于产生和使用杂交探针的组合物和方法。
本技术的实施方案提供用于产生实质上不含不想要的序列的探针(例如,缺少不想要的序列诸如重复序列的fish探针)的组合物和方法,其解决了通过提供多步骤或另外繁琐方法从探针中去除不想要的序列的现有方法中的限制。
举例说明用于产生无重复片段的ish探针的本发明的实施方案。本领域技术人员理解所公开的方法也可以应用于其他不想要的序列和其他探针应用。
fish探针中存在重复序列导致含相似重复片段的其他基因座中的背景信号。此外,这不必要地增加了fish探针的量(bulk),导致浪费材料。最后,不论重复序列,fish探针通常从直接产生自长度超过100kb的bac序列的dna制备。本文提供的方法允许选择非重复序列,对于目标基因组区域是特异的,规避了获得覆盖目标基因组区域的bac中的潜在问题并且克服了产生无重复片段的探针的现有方法中的缺点(参见例如rogan等人,genomeresearch11:1086-1094,2001;commerciallyavailableprobesfromkreatech,durham,nc;sealey等人nuc.acid.res.volume13number61985;dorman等人,nucleicacidsresearch,2013,vol.41,no.7;boyle等人,chromosomeres.2011oct;19(7):901-9;和craig等人,humgenet(1997)100:472-476)。
本文描述的探针提供了相对于现有探针的以下优点:降低来自重复序列的干扰;消除了对于人dna阻断剂的需要;更快的杂交时间;更高的杂交温度(例如,更容易去除探针且更均一的杂交温度),导致更快的杂交时间;经扩增制备(例如pcr)实现更迅速、更便宜、更可靠的制造;并且,一旦制备,模板可以在按比例扩大和制造应用中使用。
i.探针的产生
下文描述产生实质上不含不想要的序列的探针的示例性方法。举例说明用于产生ish(例如fish)探针的本发明的实施方案。本领域技术人员理解所公开的方法也可以应用于其他不想要的序列和其他探针应用。
本技术的实施方案提供用于产生和使用选择性产生或合成以排除非目标序列和/或包括目标序列的探针(例如,实质上不含重复片段的核酸探针)的组合物、试剂盒和系统。例如,在一些实施方案中,本发明提供产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp(例如,至少100、200、至少300或至少400)的目标核酸的不想要的序列(例如,无重复片段、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构或编码序列)且任选地彼此长度不超过20%不同(例如,20%或更少、10%或更少、5%或更少、4%或更少、3%或更少、3%或更少、1%或更少、或相同长度)的区域;和b)产生(例如经扩增、克隆、合成或其组合)对应于实质上不含不想要的序列的区域的多个含探针的核酸。在一些实施方案中,所述方法进一步包括以下步骤的一个或多个:c)将所述含探针的核酸片段化以产生探针;和d)进一步扩增探针的子集以产生实质上不含不想要的序列的探针(例如,缺少例如不想要的重复序列的ish探针)。在一些实施方案中,所述方法进一步包括d)根据大小分开探针的步骤。在一些实施方案中,所述分开使用层析或电泳进行。在一些实施方案中,所述方法进一步包括分离探针的子集的步骤。在一些实施方案中,所述子集基于分开的核酸的大小。在一些实施方案中,探针连接核酸衔接子。在一些实施方案中,衔接子是扩增引物。在一些实施方案中,将扩增引物功能化用于下游应用(例如通过添加标记、结合位点或限制位点)。在一些实施方案中,将探针分开和将探针子集分离。在一些实施方式中,扩增是pcr。在一些实施方案中,使用计算机软件和计算机处理器鉴定实质上不含不想要的序列的区域。在一些实施方案中,通过超声处理将含探针的核酸片段化(尽管可以使用多种其他化学、物理方法或其他方法的任一种)。在一些实施方案中,所述分开通过电泳或层析进行。在一些实施方案中,片段长度为约100至500bp,尽管也可以使用其他长度。在一些实施方案中,标记探针(例如,用荧光标记)。在一些实施方案中,探针为85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含不想要的核酸序列。
在一些实施方案中,探针长度为大约50-1000bp。例如,在一些实施方案中,探针为50-900bp、50-800bp、50-700bp、50-600bp、50-500bp、50-450bp、50-400bp、50-350bp、50-300bp、50-250bp、50-200bp、50-150bp、50-100bp、80-900bp、80-800bp、80-700bp、80-600bp、80-500bp、80-450bp、80-400bp、80-350bp、80-300bp、80-250bp、80-200bp、80-150bp、80-100bp、100-900bp、100-800bp、100-700bp、100-600bp、100-500bp、100-450bp、100-400bp、100-350bp、100-300bp、100-250bp、100-200bp、100-150bp、150-900bp、150-800bp、150-700bp、150-600bp、150-500bp、150-450bp、150-400bp、150-350bp、150-300bp、150-250bp、150-200bp、150-150bp、或80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、或150bp。在一些实施方案中,探针为85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含不想要的核酸序列。
在一些实施方案中,本发明提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)将衔接子连接至探针;和任选地e)进一步扩增探针的子集。
进一步的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;和任选地d)进一步扩增探针的子集。
另外的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含不想要的序列的区域,其中所述不想要的区域为例如长度为至少100bp的重复序列、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构或编码序列;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;和c)将所述含探针的核酸片段化以产生探针;和任选地d)进一步扩增探针的子集。
仍其他的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)通过大小分开探针;e)分离探针的子集;和任选地e)进一步扩增探针的子集。
还其他的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;d)通过大小分开探针;e)分离探针的子集,其中所述子集包含长度为80-300bp的核酸(例如,长度大约150bp);和任选地e)进一步扩增探针的子集。
另外的实施方案提供了产生针对目标核酸的探针的方法,其包括:a)鉴定目标核酸靶标的实质上不含长度为至少100bp的不想要的序列的区域;b)产生对应于所述实质上不含不想要的序列的区域的多个含探针的核酸;c)将所述含探针的核酸片段化以产生探针;任选地d)进一步扩增探针的子集产生探针组;和e)用所述探针组进行杂交测定(例如ish测定诸如fish)。
本文进一步提供通过上述方法产生的无不想要的序列的一组核酸探针(例如ish探针)和包含所述探针的试剂盒和系统。
本文另外提供进行杂交测定的方法,包括将靶核酸与通过上述方法产生的探针(例如ish探针)接触。
本文还提供任何通过上述方法产生的探针(例如ish探针)在杂交(例如ish)测定中的用途。
下文详细地描述了产生探针的示例性方法。
a.探针
本发明在一些实施方案中提供了产生探针文库的方法。在一些实施方案中,探针长度为大约100-400bp(例如,长度100-300bp)。
在一些实施方案中,产生与靶序列的不同区域互补的探针的文库。在一些实施方案中,文库中所有的探针具有相似的长度(例如,在长度1%、2%、3%、4%或5%、10%、20%之内或长度相同)。
探针可以包含任何数目的修饰的碱基、修饰的骨架(包括小沟结合剂)和标记(例如,如下文更详细所述)。此类类似物的实例包括但不限于硫代磷酸酯、氨基磷酸酯、甲基膦酸酯、手性甲基膦酸酯、2'-o-甲基核糖核苷酸和肽-核酸(pna)。
b.不想要的序列的鉴定
本发明不限于具体类型的不想要的序列。在一些实施方案中,不想要的序列例如是重复序列、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构和编码序列。在一些优选的实施方案中,将无重复片段的序列去除以优化探针结合。
在一些实施方案中,首先鉴定目标区域中基因组dna的实质上不含重复片段或其他不想要的区段。在一些实施方案中,目标区域中基因组dna的连续的无重复片段的区段通过生物信息学方法鉴定。本发明不限于具体的生物信息学方法。在一些实施方案中,采用商业软件包诸如例如repeatmaskerfunctionofucscgenomebrowser(可获自thenationalcancerinstitute'scenterforbiomedicalinformaticsandinformationtechnology),尽管也具体考虑其他商业或非商业软件包。
在一些实施方案中,使用repeat-maskerfunctionofgenomebrowser来通过将重复序列以小写字母呈现且将非重复序列以大写字母呈现来区分重复与非重复部分,并且下载序列。使用外部软件程序或序列信息的手动检查来去除序列的重复部分并以其长度次序呈现连续非重复序列的段。丢弃更短的段(通常少于300bp),并且鉴定大量更长的段使得它们组合的序列长度足以制备具有可接受的标记密度用于预期用途的探针。
c.探针的产生
鉴定不想要的序列后,设计实质上不含不想要的序列的探针。可以使用任何合适的方法来产生探针。在一些实施方案中,使用下面所述的方法扩增探针。
在一些实施方案中,合成探针文库。在一些实施方案中,合成探针包含与靶序列互补的区域和不与靶序列互补的标记区域(参见例如图1)。在一些实施方案中,标记的区域在探针文库中是相同的。在一些实施方案中,用为分支的或另一种非直链构型的标记区域产生探针。
在一些实施方案中,合成用于扩增方法或探针产生的寡核苷酸。用于寡核苷酸合成的示例性方法描述于本文。为了获得期望的寡核苷酸,将构建模块以产物序列所要求的次序顺序偶联至生长中的寡核苷酸链。自从20世纪70年代晚期,该方法已完全自动化。完成链装配后,将产物从固相释放至溶液,去保护并收集。产物通常通过高效液相层析(hplc)分离来获得高纯度的期望的寡核苷酸。通常,合成的寡核苷酸是长度约为15-25个碱基的单链dna或rna分子。
在一些实施方案中,核苷间键的形成的选择性和速率通过使用核苷的3'-o-(n,n-二异丙基)亚磷酰胺)衍生物(核苷亚磷酰胺)(其用作亚磷酸三酯法中的构建模块)来改进。为了阻止不想要的副反应,核苷中存在的所有其他官能团通过连接保护基团而变得无反应性的(受保护的)。完成寡核苷酸链装配后,去除所有保护基团以产生期望的寡核苷酸。
示例性保护基团和核苷亚磷酰胺构建模块包括但不限于酸不稳定的dmt(4,4'-二甲氧基三苯甲基)保护基团。胸腺嘧啶和尿嘧啶,分别为胸苷和尿苷的核碱基,不具有环外氨基并因此不需要任何保护。
尽管鸟苷和2'-脱氧鸟苷的核碱基不具有环外氨基,但其碱性低至一定程度使得其在偶联反应条件下不与亚磷酰胺反应。然而,衍生自n2未保护的5'-o-dmt-2'-脱氧鸟苷的亚磷酰胺在乙腈(通常用于寡核苷酸合成中的溶剂)中溶解度差。相反,相同化合物的n2保护形式充分溶解于乙腈中并因此被广泛使用。核碱基腺嘌呤和胞嘧啶携带在偶联反应条件下与活化的亚磷酰胺反应的环外氨基。通过使用合成周期中的额外步骤或可选的偶联剂和溶剂系统,使用da和dc亚磷酰胺与未保护的氨基进行寡核苷酸链装配。在一些实施方案中,核苷中的环外氨基在寡核苷酸链装配的整个长度上保持永久受保护。
环外氨基的保护通常与5'-羟基的保护是正交的(orthogonal),因为后者在每一合成周期结束时去除。最容易实现且因此最广泛接受的是其中环外氨基携带碱不稳定保护的策略。最经常地,使用两种保护方案。
在一些实施方案中,对于a、da、c和dc使用bz(苯甲酰基)保护,而用异丁酰基保护g和dg。更近期,经常用ac(乙酰基)来保护c和dc。
在第二种、温和的保护方案中,用异丁酰基或苯氧基乙酰基(pac)保护a和da。c和dc携带乙酰基保护,和g和dg用4-异丙基苯氧基乙酰基(ipr-pac)或二甲基亚胺甲基氨(dmf)基团保护。温和的保护基团比标准保护基团更容易去除。然而,携带这些基团的亚磷酰胺当在溶液中储存时更不稳定。
在一些实施方案中,亚磷酸基团被碱不稳定的2-氰基乙基保护。一旦亚磷酰胺已经与固体支持物结合的寡核苷酸偶联并且亚磷酸部分已经转化为p(v)种类,存在磷酸保护对于成功进行进一步的偶联反应不再是强制性的。
非核苷亚磷酰胺是设计用于在合成的寡核苷酸的末端或在序列中间的核苷酸残基之间引入不同官能度的亚磷酰胺试剂。为了被引入到序列内部,非核苷改性剂需要具有至少两个羟基,其中一个通常用dmt基团保护而另一个携带反应性亚磷酰胺部分。
使用非核苷亚磷酰胺来引入在天然核苷中不存在的或可以使用更简单的化学设计更容易地引入的期望的基团。
寡核苷酸合成通过逐步添加核苷酸残基至生长中的链的5'末端来进行直至装配完期望的序列。每一添加称为合成周期并且由四个化学反应组成:
步骤1:去阻断(脱三苯甲基化)
用酸溶液诸如2%三氯乙酸(tca)或3%二氯乙酸(dca)在惰性溶剂(二氯甲烷或甲苯)中去除dmt基团。将形成的橙色的dmt阳离子洗去;该步骤导致携带游离5'末端羟基的固体支持物结合的寡核苷酸前体。进行脱三苯甲基化持续延长的时间或用比推荐的酸溶液更强的来进行导致固体支持物结合的寡核苷酸的脱嘌呤并因此降低了期望的全长产物的产率。
乙腈中的核苷亚磷酰胺(或若干亚磷酰胺的混合物)的溶液随后通过酸性唑(acidicazole)催化剂1h-四唑、2-乙基硫代四唑、2-苄基硫代四唑、4,5-二氰基咪唑或多种类似化合物来活化。混合通常非常短暂且在寡核苷酸合成仪的流体线中发生,同时组分被递送至含有固体支持物的反应器。相对于支持物结合材料1.5-20倍过量的活化的亚磷酰胺随后与起始固体支持物接触(第一偶联)或与支持物结合的寡核苷酸前体接触(随后偶联),所述寡核苷酸前体的5'-羟基与进入的核苷酸亚磷酰胺的活化的亚磷酰胺部分反应以形成亚磷酸三酯键。2'-脱氧核苷亚磷酰胺的偶联非常迅速且对于其完成在小规模上需要约20秒。相反,位阻的2'-o-保护的核糖核苷亚磷酰胺利用更长时间来以高产率偶联。反应对于水的存在也是高度敏感的,特别是当使用亚磷酰胺的稀释溶液时,且通常在无水的乙腈中进行。通常,合成的规模越大,使用亚磷酰胺的过量越低且浓度越高。相反,活化剂的浓度主要由其在乙腈中的溶解度决定且与合成规模无关。完成偶联后,通过洗涤去除任何未结合的试剂和副产物。
封端步骤在亚磷酰胺方法中通过用乙酸酐和1-甲基咪唑或更少见地dmap的混合物作为催化剂处理固体支持物结合的材料来进行,有两个目的。
完成偶联反应后,小百分比的固体支持物结合的5'-oh基团(0.1至1%)保持未反应且需要从进一步的链延长中永久阻断以防止通常称为(n-1)短聚物(shortmer)的具有内部碱基缺失的寡核苷酸的形成。未反应的5'-羟基在很大程度上通过封端混合物乙酰化。
还已经报道用1h-四唑活化的亚磷酰胺在较小程度上与鸟苷的o6位反应。用i2/水氧化后,这一副产物可能经o6-n7迁移经历脱嘌呤。因此形成的无嘌呤位点在碱性条件下(见下文)在寡核苷酸的最终去保护过程中容易地被切割以产生两个较短的寡核苷酸,因此降低了全长产物的产率。o6修饰通过用封端试剂处理容易地被去除,只要在用i2/水氧化前进行封端步骤。
寡核苷酸硫代磷酸酯(ops)的合成不涉及用i2/水氧化,并且分别不经受上述副反应。另一方面,如果封端步骤在硫化前进行,则固体支持物包含在封端步骤后留下的残留的乙酸酐和n-甲基咪唑。封端混合物干扰硫转移反应,其导致磷酸三酯核苷内键的大量形成替代了期望的ps三酯。因此,对于ops的合成,建议在封端步骤前进行硫化步骤。
新形成的三配位的亚硫酸三酯键不是天然的且在寡核苷酸合成的条件下具有有限的稳定性。用碘和水在弱碱(吡啶、二甲基吡啶或三甲吡啶)存在的情况下处理支持物结合的材料氧化亚磷酸三酯为四配位的磷酸三酯,其为天然存在的磷酸二酯核苷间键的受保护的前体。氧化可以在无水条件下使用叔丁基过氧化氢或更有效地使用(1s)-(+)-(10-樟脑磺酰)-氧氮环丙烷(cso)进行。氧化步骤用硫化步骤替代以获得寡核苷酸硫代磷酸酯。在后一种情况中,硫化步骤最好在封端前进行。
在固相合成中,装配的寡核苷酸经其3'末端羟基共价结合至固体支持物材料并且在链装配整个过程中保持与其连接。固体支持物包含在柱内,所述柱的直径取决于合成规模且可以在0.05ml至若干升之间改变。绝大多数寡核苷酸以40nmol至1μmol范围的小规模合成。更近期,其中固体支持物包含在多孔板(最常见地,96或384孔/板)的孔内的高通量寡核苷酸合成成为以小规模平行合成寡核苷酸的方法选择。在链装配结束时,寡核苷酸从固体支持物上释放并从柱或孔上洗脱。
与有机固相合成和肽合成不同,寡核苷酸的合成在无膨胀或低膨胀的固体支持物上进行最好。两种最常用的固相材料是可控孔度玻璃(cpg)和大孔聚苯乙烯(mpps)。
cpg通常由其孔径定义。在寡核苷酸化学中,使用500、1000、1500、2000和3000å的孔径以允许分别制备约50、80、100、150和200聚体(mer)的寡核苷酸。为了制备适合于进一步加工的天然cpg,将材料表面用(3-氨基丙基)三乙氧基硅烷处理以产生氨基丙基cpg。氨基丙基臂可以进一步延伸以产生长链氨基烷基(lcaa)cpg。随后将氨基用作适合于寡核苷酸合成的接头的锚定点(见下文)。
适合于寡核苷酸合成的mpps是低膨胀的、高度交联的聚苯乙烯,其通过将二乙烯基苯(最少60%)、苯乙烯和4-氯甲基苯乙烯在成孔剂(porogeneousagent)存在的情况下聚合来获得。将获得的大孔氯甲基mpps转化为氨基甲基mpps。
为了制备适合于寡核苷酸合成的固体支持物材料,将非核苷接头或核苷琥珀酸酯共价连接至氨基丙基cpg、lcaacpg或氨基甲基mpps中的反应性氨基。剩余未反应的氨基用乙酸酐封端。通常,使用固体支持物的三种概念上不同的基团。
在更近期、更方便且更广为使用的方法中,合成开始于通用支持物,其中非核苷接头连接至固体支持物材料。3'末端核苷残基各自的亚磷酰胺在寡核苷酸链装配的第一合成周期中使用标准方案偶联至通用固体支持物。链装配随后持续直至完成,之后将固体支持物结合的寡核苷酸去保护。通用固体支持物的特征性特点在于寡核苷酸的释放通过连接3'末端核苷酸残基的3'-o与通用接头的p-o键的水解切割发生,如方案6中所示。这一方法的主要优点是使用相同的固体支持物,而与待合成的寡核苷酸序列无关。对于从装配的寡核苷酸中完全去除接头和3'末端磷酸酯,固体支持物1和若干相似的固体支持物需要气态氨、氢氧化铵水溶液、甲胺水溶液、或其混合物,且是可商购的。固体支持物利用无水甲醇中的氨溶液并且是可商购的。
通常,3'末端核苷残基的3'-羟基经(最常见地)如化合物3中的3'-o-琥珀酰臂连接至固体支持物。寡核苷酸链装配起始于各自对于从3'末端第二的核苷酸残基的亚磷酰胺构建模块的偶联。核苷固体支持物上合成的寡核苷酸中3'末端羟基在稍微比适用于通用固体支持物的条件更温和的条件下进行去保护。然而,核苷固体支持物必须以序列特异性方式选择这一事实降低了整个合成方法的通量且增加了人为错误的可能性。
寡核苷酸硫代磷酸酯(ops)是修饰的寡核苷酸,其中磷酸部分中的一个氧原子被硫所取代。只有在非桥接位置具有硫的硫代磷酸酯是广泛使用的且可以商购的。用硫取代非桥接的氧产生在磷处的新的手性中心。在二核苷酸的简单情况中,这导致形成sp-和rp-二核苷单硫代磷酸酯的非对映体对。在其中所有(n-1)核苷间键是硫代磷酸酯键的n-聚体寡核苷酸中,非对映体的数目m计算为m=2(n–1)。作为核酸的非天然类似物,ops对于通过核酸酶(通过断裂磷酸二酯部分的桥连p-o键来破坏核酸的酶类)的水解基本上更稳定。该性质决定了在其中广泛暴露于核酸酶是不可避免的体外和体内应用中ops作为反义寡核苷酸的用途。类似地,为了提高sirna的稳定性,通常在有义链和反义链的3'-末端引入至少一个硫代磷酸酯键。在手性纯ops中,全sp非对映体比其全部rp类似物对于酶促降解更加稳定。然而,手性纯ops的制备仍然是合成挑战。在实验室实践中,通常使用ops的非对映体的混合物。
ops的合成与天然寡核苷酸的合成非常相似。不同之处在于氧化步骤被硫转移反应(硫化)代替,并且封端步骤在硫化之后进行。在许多报道的能够有效转移硫的试剂中,只有三种可商购:
3-(二甲氨基亚甲基)氨基)-3h-1,2,4-二噻唑-3-硫酮,ddtt(3)提供快速的硫化动力学和溶液中的高稳定性。也称为beaucage试剂的3h-1,2-苯并二硫醇-3-酮1,1-二氧化物(4)在乙腈中显示更好的溶解度和较短的反应时间。然而,试剂在溶液中的稳定性有限,并且在硫化rna键中效率较低。
n,n,n'n'-二硫化四乙基秋兰姆(tetd)可溶于乙腈且可商购。然而,核苷间dna键与tetd的硫化反应需要15分钟。
在过去,在溶液中或固相上手动进行寡核苷酸合成。使用其形状类似于低压层析柱的微型玻璃柱或装配有多孔过滤器的注射器作为固相的容器进行固相合成。目前,使用计算机控制仪器(寡核苷酸合成仪)自动进行固相寡核苷酸合成且技术上以柱、多孔板和阵列格式实现。柱格式最适合于不需要高通量的研究和大规模应用。多孔板格式专门设计用于小规模的高通量合成,以满足工业和学术界对合成寡核苷酸不断增长的需求。许多用于小规模合成和中等至大规模合成的寡核苷酸合成仪是可商购的。
下面描述扩增方法,尽管可以使用其它方法。接下来,设计扩增(例如pcr)引物以从使用生物信息学方法鉴定的最长的无重复片段的区段中扩增序列的段。在一些实施方案中,使用基因组或基因组衍生的bacdna作为扩增模板来扩增这些区段。在一些实施方案中,(例如,在较长的段的情况下)使用多个重叠引物组。
核酸扩增技术的说明性非限制性实例包括但不限于聚合酶链反应(pcr)、逆转录聚合酶链反应(rt-pcr)、转录介导的扩增(tma)、连接酶链反应(lcr)、链置换扩增(sda)和基于核酸序列的扩增(nasba)。
通常,扩增方法使用dna聚合酶、引物和dntp。示例性dna聚合酶包括但不限于phi29dna聚合酶、taqdna聚合酶、dna聚合酶i、t7dna聚合酶、t7dna聚合酶、t4dna聚合酶、pfudna聚合酶和bsmdna聚合酶。
聚合酶链反应(美国专利号4,683,195、4,683,202、4,800,159和4,965,188,其各自以其整体通过引用并入本文),通常称为pcr,使用多个循环的变性、引物对与相对链的退火、和引物延伸以指数增加靶核酸序列的拷贝数。在称为rt-pcr的变化中,逆转录酶(rt)用于从mrna制备互补dna(cdna),然后通过pcr扩增cdna以产生多个dna拷贝。对于pcr的其他各种改变,参见例如美国专利号4,683,195、4,683,202和4,800,159;mullis等人,meth.enzymol.155:335(1987);和murakawa等人,dna7:287(1988),其各自以其整体通过引用并入本文。
转录介导的扩增(美国专利号5,480,784和5,399,491,其各自以其整体通过引用并入本文),通常称为tma,在基本上恒定的温度、离子强度和ph的条件下自催化合成靶核酸序列的多个拷贝,其中靶序列的多个rna拷贝自催化产生额外的拷贝。参见例如美国专利号5,399,491和5,824,518,其各自以其整体通过引用并入本文。在美国公开号20060046265(以其整体通过引用并入本文)中描述的变化中,tma任选地并入使用阻断部分、终止部分和其它修饰部分以提高tma方法的灵敏度和精度。
连接酶链反应(weiss,r.,science254:1292(1991),以其整体通过引用并入本文),通常称为lcr,使用两组与靶核酸的邻近区域杂交的互补dna寡核苷酸。通过dna连接酶在热变性、杂交和连接的重复循环中共价连接dna寡核苷酸以产生可检测的双链连接的寡核苷酸产物。
链置换扩增(walker,g.等人,proc.natl.acad.sci.usa89:392-396(1992);美国专利号5,270,184和5,455,166,其各自以其整体通过引用并入本文),通常称为sda,使用引物序列对与靶序列的相对链的退火、在dntp存在下的引物延伸的循环以产生双链体半硫代磷酸化的引物延伸产物,半修饰的限制性内切核酸酶识别位点的内切核酸酶介导的切口,以及从切口的3'末端的聚合酶介导的引物延伸以置换现有的链并产生用于下一轮引物退火、切口和链置换的链,导致产物的几何扩增。嗜热sda(tsda)在基本相同的方法中在较高温度下使用嗜热内切核酸酶和聚合酶(欧洲专利号0684315)。
其他扩增方法包括,例如:基于核酸序列的扩增(美国专利号5,130,238,以其整体通过引用并入本文),通常称为nasba;使用rna复制酶扩增探针分子自身的扩增方法(lizardi等人,biotechnol.6:1197(1988),以其整体通过引用并入本文),通常称为qβ复制酶;基于转录的扩增方法(kwoh等人,proc.natl.acad.sci.usa86:1173(1989));和,自维持序列复制(guatelli等人,proc.natl.acad.sci.usa87:1874(1990),其各自以其整体通过引用并入本文)。对于已知扩增方法的进一步讨论,参见persing,davidh.,“invitronucleicacidamplificationtechniques”于diagnosticmedicalmicrobiology:principlesandapplications(persing等人编辑),第51-87页(americansocietyformicrobiology,washington,dc(1993))。
在一些实施方案中,扩增是等温扩增。在一些实施方案中,扩增方法是固相扩增、群落扩增(polonyamplification)、菌落扩增、乳液pcr、珠粒rca、表面rca、表面sda等,如将由本领域技术人员所认可。在一些实施方案中,使用导致扩增溶液中的游离dna分子或通过dna分子的仅一个末端拴系至合适的基材的dna分子的扩增方法。在一些实施方案中,使用依赖于桥式pcr的方法,其中两条pcr引物均连接至表面(参见例如wo2000/018957,u.s.7,972,820;7,790,418和adessi等人,nucleicacidsresearch(2000):28(20):e87;其各自以其整体通过引用并入本文)。在一些情况下,本发明的方法可以产生“聚合酶菌落技术”或“群落(polony)”,指保持相同扩增子的空间聚类的多路扩增(参见harvardmoleculartechnologygroupandlippercenterforcomputationalgenetics网站)。这例如包括原位群落(mitra和church,nucleicacidresearch27,e34,dec.15,1999)、原位滚环扩增(rca)(lizardi等人,naturegenetics19,225,july1998)、桥式pcr(美国专利号5,641,658),皮滴定pcr(picotiterpcr)(leamon等人,electrophoresis24,3769,november2003)和乳液pcr(dressman等人,pnas100,8817,jul.22,2003)。
适合用于本发明的实施方案的核酸聚合酶的实例包括但不限于dna聚合酶(klenow片段,t4dna聚合酶)、热稳定的dna聚合酶(perlerf.b.等人,adv.proteinchem.1996,48:377-435),其鉴定并克隆了多种热稳定细菌(诸如taq、vent、pfu、tfldna聚合酶)及其遗传学修饰的衍生物(taqgold、ventexo、pfuexo)。用于菌落引物延伸的核酸聚合酶优选在引物和模板杂交结果足够特异以避免模板的不完全或假扩增的温度下是稳定的。
扩增溶液优选含有脱氧核糖核苷三磷酸,例如datp、dttp、dctp、dgtp,其为天然或非天然存在的,例如用荧光或放射性基团修饰的。为了提高核酸的可检测性和/或功能多样性,已经开发了各种各样的合成修饰的核酸用于化学和生物学方法。这些功能化/修饰的分子(例如核苷酸类似物)可以与天然聚合酶完全相容,保持天然对应物的碱基配对和复制性质,如最近所综述的(thumo等人,angew.chem.int.ed.2001,40(21):3990-3993)。
因此,将扩增溶液的其他组分加入到核酸聚合酶的选择中,并且它们基本上对应于本领域已知的有效支持每种聚合酶活性的化合物。化合物如二甲基亚砜(dmso)、牛血清白蛋白(bsa)、聚乙二醇(peg)、甜菜碱、tritonx-100或mgcl2的浓度由于对于产生最佳扩增是重要的,因此在现有技术中是众所周知的,且因此操作人员可以基于下面给出的实例容易地调整用于本发明方法的此类浓度。
d.片段化
在一些实施方案中,扩增前和扩增后,随后将dna片段化(例如,通过超声处理或其他合适的方法诸如dna酶i)至大约50-5000bp范围的长度(例如50-4000、50-3000、50-2500、50-2000、50-1500、50-1000、100-5000、100-4000、1000-3000、100-2500、100-2000、100-1500、100-1000或100-500bp),并且所得的无重复片段的dna文库连接(例如经连接、化学、延伸反应等)至衔接子。在一些优选的实施方案中,探针为大约150bp(例如,50-900bp、50-800bp、50-700bp、50-600bp、50-500bp、50-450bp、50-400bp、50-350bp、50-300bp、50-250bp、50-200bp、50-150bp、50-100bp、80-900bp、80-800bp、80-700bp、80-600bp、80-500bp、80-450bp、80-400bp、80-350bp、80-300bp、80-250bp、80-200bp、80-150bp、80-100bp、100-900bp、100-800bp、100-700bp、100-600bp、100-500bp、100-450bp、100-400bp、100-350bp、100-300bp、100-250bp、100-200bp、100-150bp、150-900bp、150-800bp、150-700bp、150-600bp、150-500bp、150-450bp、150-400bp、150-350bp、150-300bp、150-250bp、150-200bp、150-150bp、或80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、或150bp等)。在一些实施方案中,随后将衔接的文库分级(例如通过电泳、层析、或其他分开方法)以产生含有多种片段大小的所有所选序列的库。将对应于期望的片段大小的级分(例如凝胶片)分离。
f.扩增
在一些实施方案中,将所需大小的级分用作在准备扩增条件下用对应于衔接子的扩增(例如,本文所述的pcr或另一种方法)引物的模板。
所有选择的无重复片段的序列都代表在扩增的文库中。然后将探针(例如,在片段化或任选的进一步扩增后分离的)标记(例如,用荧光标记、生物素、量子点标记或用于比色或银染检测的标记)用于使用(例如,作为fish试剂)。
在一些实施方案中,将探针文库克隆到表达载体中(例如,每个载体一个或多个探针)。在一些实施方案中,这些表达载体可用于将来生成文库(例如通过扩增或表达)。
ii.探针的用途
本文所述的探针可用于多种诊断、研究、临床和筛选应用中。所描述的核酸杂交探针广泛用于通过使用核酸杂交探针实现的所有形式的核酸检测。核酸杂交探针用于检测溶液中的核酸序列靶标或与固定的支持物结合的核酸序列靶标。组合物和方法可用于检测溶液中核酸序列靶标的应用实例包括pcr、实时pcr、定量pcr、pna钳介导pcr和数字pcr。组合物和方法可用于检测固定于固体支持物上的核酸序列靶标的应用实例包括northern印迹、southern印迹、斑点印迹,槽印迹、微阵列、基于粒子的测定、原位杂交测定(ish),诸如例如色原原位杂交(cish)、rna原位杂交(rish)、快速fish、银原位杂交(sish)和fish测定。此类应用适用于许多领域,包括医学诊断、分子医学、法医学、标本和生物编目、以及微生物病原体流行病学。
本发明不限于特定的靶标。本文描述的组合物和方法可用于检测多种靶核酸(例如人或哺乳动物基因组核酸),细菌靶核酸,病毒靶核酸等。
探针也可以用作固定在固体表面(例如硝酸纤维素)上的分离的核酸,如在acgh中。在一些实施方案中,探针可以是例如在wo96/17958中所述的核酸阵列的成员,wo96/17958以其整体通过引用并入本文并且具体涉及其对阵列cgh的描述。能够产生高密度阵列的技术是众所周知的(参见例如fodor等人science767-773(1991)和美国专利号5,143,854),其两者对于其描述均通过引用并入本文。
利用核酸杂交探针的详细描述在下文对于fish应用进行呈现,尽管探针也可用于其它应用。
fish和其他原位杂交方法可以在各种样品类型上进行。实例包括但不限于福尔马林固定石蜡包埋(ffpe)组织)、新鲜组织、冷冻组织、细胞(例如真核或原核细胞);其使用任何合适的固定剂制备。在一些实施方案中,采用印片制备(touchprep)或刷涂(brushing)(参见例如smoczynski等人,gastrointestendosc.2012jan;75(1):65-73)。
印片制备样本通过涂抹或压在载玻片上,向组织施加压力并在冷却温度下固定在乙醇中而产生。在一个具体实施方案中,手术提取组织并通过对肿瘤组织施加相对弱的压力并对正常组织施加相对强的压力将其涂抹在载玻片上,然后在约4℃下固定在约100%乙醇中约10分钟。另一个具体实施方案中,将通过本发明的方法分析的样品最初在液氮中冷冻并储存在约-80℃。
对于典型的ish应用,以下内容代表典型的程序。收获样本的细胞,洗涤并沉淀。沉淀物的细胞通常在磷酸缓冲盐水(pbs)中洗涤。将细胞悬浮在pbs中并通过离心再收集。细胞可以例如固定在酸醇溶液、酸丙酮溶液、或醛诸如甲醛、低聚甲醛和戊二醛中。例如,含有分别为3:1比例的甲醇和冰醋酸的固定液可用作固定液。也可以使用中性缓冲的福尔马林溶液,并且包括在磷酸钠的水溶液中大约1%-10%的37-40%甲醛。含有细胞的载玻片可以通过去除大部分固定液,保留悬浮在仅部分溶液中的浓缩细胞来制备。
将细胞悬液施加到载玻片上使得细胞不会在载玻片上重叠。细胞密度可以通过光或相差显微镜来测量。这些孔中的细胞密度随后用相差显微镜来评估。如果含有最大体积的细胞悬液的孔不具有足够的细胞,则将细胞悬液浓缩并置于另一孔中。
在原位杂交前,将染色体探针和包含在细胞内的染色体dna各自变性。变性过程以几种方式进行。例如,可以用具有升高的ph、具有升高的温度(例如,温度从约70℃至约95℃)、或具有有机溶剂如甲酰胺、碳酸亚乙酯和四烷基卤化铵、或其组合的缓冲溶液实现变性。例如,染色体dna可以通过高于70℃的温度(例如约73℃)和含有70%甲酰胺和2×ssc(0.3m氯化钠和0.03m柠檬酸钠)的变性缓冲液的组合而变性。变性条件通常这样确立使得保留细胞形态。染色体探针可以通过热来变性。例如,可以将探针加热至约73℃持续约五分钟。
在去除变性化学品或条件后,探针在杂交条件下与染色体dna退火。“杂交条件”是促进探针和核酸序列靶标之间退火的条件。杂交条件取决于探针的浓度、碱基组成、复杂性和长度以及盐浓度、温度和孵育时间而有所不同。探针的浓度越大,形成杂交物的概率就越大。例如,原位杂交通常在含有1-2×ssc、50%甲酰胺和封闭dna的杂交缓冲液中进行,以抑制非特异性杂交。通常,如上所述的杂交条件包括约25℃至约55℃的温度和约0.5小时至约96小时的孵育时间。更具体地,可以在约37℃至约40℃进行约2至约16小时的杂交。
染色体探针与目标区域外的dna的非特异性结合可以通过一系列洗涤来去除。每次洗涤中盐的温度和浓度取决于所需的严格性。例如,对于高严格条件,洗涤可以在约65℃至约80℃下进行,使用0.2×ssc至约2×ssc和约0.1%至约1%的非离子型去污剂如nonidetp-40(np40)或其他合适的表面活性剂。通过降低洗涤温度或通过增加洗涤中的盐浓度可以降低严格性。
含有样品的载玻片通常在2×ssc中在37℃下孵育10-30分钟。然后将载玻片在0.2mg/ml胃蛋白酶中于37℃温育20分钟。随后将载玻片在pbs中在室温下洗涤两次2分钟。细胞在2.5%中性缓冲的福尔马林中在室温下固定5分钟。随后将载玻片在pbs中在室温下洗涤两次2分钟。通过在室温下在70%、85%和100%乙醇的溶液中连续接触1分钟,使载玻片脱水。之后立即使用载玻片或在室温下在黑暗中储存。
可以使用hybrite方法或常规方法进行杂交。在hybrite方法中,使用来自abbottmolecular(downersgrove,il)的hybritetm系统。
用于探针与其核酸靶标特异性杂交的条件通常包括在给定杂交程序中可用于产生特异性杂交物的条件的组合,该条件可以由本领域技术人员容易地确定。这些条件通常涉及受控的温度、液相和染色体探针与靶标之间的接触。杂交条件取决于许多因素而不同,包括探针浓度、靶标长度、靶标和探针g-c含量、溶剂组成、温度和培养持续时间。至少一个变性步骤可能在探针与靶标接触之前。或者,探针和核酸靶标两者可以在彼此接触的同时经历变性条件,或者随后探针与生物样品接触。杂交可以通过随后在约25至约55℃的温度范围下将探针/样品温育在例如2-4×ssc和甲酰胺的约50:50体积比混合物的液相中持续说明性地在约0.5-约96小时范围内的时间,或者更优选在约32-约40℃的温度下持续约2-约16小时范围的时间来实现。为了增加特异性,使用封闭剂诸如美国专利号5,756,696中所述的未标记的封闭核酸(所述专利内容以其整体通过引用并入本文,并且具体涉及封闭核酸的使用的描述),可以与本发明的方法联合使用。如本领域技术人员显而易见的,其他条件可以容易地用于将探针与存在于样品中的其核酸靶标特异性杂交。
在合适的孵育期完成后,染色体探针与样品dna的非特异性结合可以通过一系列洗涤来除去。适当地选择温度和盐浓度以达到所需的严格性。所需的严格性水平取决于与基因组序列相关的特异性探针序列的复杂度,并且可以通过将探针与已知遗传组成的样品系统杂交来确定。通常,高严格性洗涤可以在约65至约80℃范围内的温度下进行,用约0.2×至约2×ssc和约0.1%至约1%的非离子型去污剂例如nonidetp-40(np40)。如果需要较低的严格性洗涤,洗涤可以在较低温度且增加的盐浓度下进行。
可以用可检测标记直接标记染色体探针。可检测标记的实例包括荧光团,例如吸收光后发荧光的有机分子以及放射性同位素,例如32p和3h。荧光团可以在通过用标准技术如切口平移、随机引发和pcr标记将标记的核苷酸掺入探针中共价连接至核苷酸后而被直接标记。或者,探针内的脱氧胞苷核苷酸可以用接头转氨化。荧光团可以随后共价连接至转氨化的脱氧胞苷核苷酸。参见例如美国专利号5,491,224,bittner等人,其通过引用并入本文。可用的探针标记技术描述于molecularcytogenetics:protocolsandapplications,y.-s.fan,编辑,第2章,“labelingfluorescenceinsituhybridizationprobesforgenomictargets”,l.morrison等人,p.21-40,humanapress,©2002(下文引用为“morrison-2002”),其通过引用并入本文。
荧光团与核酸探针的连接是本领域中熟知的且可以通过任何可用的方法完成。荧光团可以共价连接到特定的核苷酸,例如,并且使用标准技术(例如切口平移,随机引发,pcr标记等)将标记的核苷酸掺入探针中。或者,荧光团可以经由接头共价连接到已经被转氨化的探针的脱氧胞苷核苷酸上。用于标记探针的方法描述于美国专利号5,491,224和molecularcytogenetics:protocolsandapplications(2002),y.-s.fan,编辑,第2章“labelingfluorescenceinsituhybridizationprobesforgenomictargets,”l.morrison等人,p.21-40,humanapress,其两者均对于其关于标记探针的描述通过引用并入本文。
可用于标记探针的示例性荧光团包括texasred(molecularprobes,inc.,eugene,oreg.)、cascadeblueaectylazide(molecularprobes,inc.,eugene,oreg.)、spectrumorangetm(abbottmolecular,desplaines,ill.)和spectrumgoldtm(abbottmolecular)。
可用于本文描述的方法中的荧光团的另外实例是:7-氨基-4-甲基香豆素-3-乙酸(amca);5-(和-6)-羧基-x-罗丹明、丽丝胺罗丹明b、5-(和-6)-羧基荧光素;荧光素-5-异硫氰酸酯(fitc);7-二乙基氨基香豆素-3-羧酸、四甲基-罗丹明-5-(和-6)-异硫氰酸酯;5-(和-6)-羧基四甲基罗丹明;7-羟基香豆素-3-羧酸;6-[荧光素5-(和-6)-酰胺基]己酸;n-(4,4-二氟-5,7-二甲基-4-硼-3a,4a二氮杂-3-引达省丙酸(indacenepropionicacid);曙红-5-异硫氰酸酯;赤藓红-5-异硫氰酸酯;5-(和-6)-羧基罗丹明6g;和cascadesblueaectylazide(molecularprobes,inc.,eugene,oreg.)。
可以使用荧光显微镜和每种荧光团的适当滤光片或通过使用双重或三重带通滤光片组来观察多种荧光团以观察探针。参见例如美国专利号5,776,688,bittner等人,其通过引用并入本文。可以使用任何合适的显微镜成像方法来显现杂交探针,包括自动数字成像系统,例如可从metasystems或appliedimaging获得的那些。或者,可以使用技术诸如流式细胞术来检查染色体探针的杂交模式。
也可以通过本领域熟知的方法例如用生物素或地高辛间接地标记探针。然而,随后使用二级检测分子或进一步处理来可视化标记的探针。例如,用生物素标记的探针可以通过与可检测标记物例如荧光团缀合的抗生物素蛋白来检测。此外,抗生物素蛋白可以与酶标记物如碱性磷酸酶或辣根过氧化物酶缀合。可以使用酶的底物在标准比色反应中检测此类酶标记物。碱性磷酸酶的底物包括5-溴-4-氯-3-吲哚基磷酸酯和硝基蓝四唑鎓。二氨基苯甲酸酯可用作辣根过氧化物酶的底物。杂交的生物素或其他间接标记的探针的荧光检测可以通过使用市售的酪胺放大系统来实现。
本领域技术人员将认识到可以使用其它试剂或染料代替荧光团作为含标记的部分。可以附着于探针的合适的标记包括但不限于放射性同位素、荧光团、发色团、质量标签、电子致密颗粒、磁性颗粒、自旋标记、发射发光的分子、电化学活性分子、酶、辅因子和酶底物。发光剂包括例如放射性发光、化学发光、生物发光和含磷光标记的部分。或者,可以使用通过间接方法可视化的检测部分。例如,可以使用本领域已知的常规方法用生物素或地高辛标记探针,然后进一步处理用于检测。含有生物素的探针的可视化可以通过与可检测标记物缀合的抗生物素蛋白的随后结合来实现。可检测标记物可以是荧光团,在这种情况下可以如上文针对ish所述实现探针的可视化和辨别。
在一些实施方案中,探针被设计成具有在整个核酸中以共同间隔放置的标记(例如每3、4、5、6、7、8、9、10、11或12个一个标记组)。
在一些实施方案中,探针文库包含具有不同可检测标记(例如,不同颜色的荧光信号)的探针。
与目标区域杂交的探针可以可选地通过标记部分与合适底物的酶反应用于生产不溶性颜色产物来显现。可以通过随后与缀合至碱性磷酸酶(ap)或辣根过氧化物酶(hrp)的抗生物素蛋白和合适底物孵育来检测组内的含生物素的探针。5-溴-4-氯-3-吲哚基磷酸酯和硝基蓝四唑鎓(nbt)用作碱性磷酸酶的底物,而二氨基联苯胺作为hrp的底物。
在使用荧光团标记的探针或探针组合物的实施方案中,检测方法可以涉及荧光显微镜、流式细胞术或用于确定探针杂交的其它方法。任何合适的显微镜成像方法可以结合本发明的方法使用用于观察多种荧光团。在使用荧光显微镜的情况下,可以在适合于激发每种荧光团的光下并且使用合适的一种或多种滤光片来观察杂交样品。可选地可以使用自动数字成像系统诸如metasystems、bioview或appliedimaging系统。
在阵列cgh中,探针未被标记,而是固定在基底上的不同位置,如wo96/17958中所述。在这种情况下,探针通常被称为“靶核酸”。样品核酸通常被标记以允许检测杂交复合物。用于杂交的样品核酸可以在杂交反应之前被可检测地标记。或者,可以选择结合杂交产物的可检测标记。在双色或多色acgh中,靶核酸阵列同时或连续地与不同标记的核酸的两个或多个集合杂交。例如,样品核酸和参考核酸各自用独立且可区分的标记进行标记。可以检测每个靶核酸斑点处的每个信号的强度差异作为拷贝数差异的指示。尽管任何合适的可检测标记可用于acgh,但荧光标记通常是最方便的。
在wo93/18186中描述了可视化信号的示例性方法,其对于该描述通过引用并入本文。为了便于显示结果和提高检测荧光强度微小差异的灵敏度,可以使用数字图像分析系统。示例性系统是quips(定量图像处理系统的首字母缩略词),其是基于配备有自动化阶段、焦点控制和滤光轮的标准荧光显微镜的自动图像分析系统(ludlelectronicproducts,ltd.,hawthorne,ny.)。滤光轮固定在显微镜的荧光激发路径上用于选择激发波长。分色镜中的特殊滤光片(chromatechnology,brattleboro,vt.)允许激发多种染料而无图像配准位移。显微镜有两个相机端口,其中一个具有加强的ccd相机(quantexcorp.,sunnyvale,calif.)用于灵敏的高速视频图像显示,其用于在载玻片上找到兴趣区域以及用于聚焦。另一相机端口具有冷却ccd相机(型号200,来自photometricsltd.,tucson,ariz.),其以高分辨率和灵敏度用于实际图像采集。冷却ccd相机通过vme总线连接到sun4/330工作站(sunmicrosystems,inc.,mountainview,calif.)。使用图像处理软件包scil-image(delftcenterforimageprocessing,delft,netherlands)控制多色图像的整个采集。
在一些实施方案中,本公开提供用于扩增和/或分析核酸的试剂盒和系统。在一些实施方案中,试剂盒包括用于分析和检测拷贝数或基因表达变化必需的、足够的或可用的试剂(例如引物、探针、锚(anchors)、固体支持物、试剂、对照、说明书等)。例如,在一些实施方案中,试剂盒包含用于扩增和测序兴趣区域和对照区域的引物和锚。在一些实施方案中,试剂盒包括分析软件(例如,以分析测序数据)。
在一些实施方案中,试剂盒包含一个或多个含有试剂、引物、探针、锚、固体支持物、缓冲液等的容器。在一些实施方案中,试剂盒的每个组分包装在单独的容器中。在一些实施方案中,容器被包装和/或运输在相同的试剂盒或盒中以一起使用。在一些实施方案中,试剂盒的一种或多种组分被分开运输和/或包装。
可适用于现场即时(pointofcare)测定系统(包括abbott'spointofcare(i-stattm)电化学免疫测定系统免疫传感器)或对其优化的测定和试剂盒以及制造它们和在一次性测试装置中操作它们的方法例如描述于美国专利号5,063,081和公开的美国专利申请号20030170881、20040018577、20050054078和20060160164中(对于其关于所述内容的教导通过引用并入本文)。
在一些实施方案中,系统包括自动化样品和试剂处理装置(例如机器人)。
实验
实施例1
探针的产生
使用ucsc基因组浏览器来鉴定坐标为hg18_dna范围chr17:35004678-35230380的her2基因座区域中的序列。将该序列下载,编码,使得由repeatmasker鉴定的区域被掩蔽为小写字母。将225703个碱基的序列用软件程序进行处理,该软件程序删除了具有多于连续3个小写字母的所有序列,返回无重复片段的序列的段及其在原始序列中的位置(图2)。
这些序列的大小从非常小到5615个碱基。仅选择最长的片段来生成序列库。1200bp的最小长度截止值产生35个序列,其包含76904bp的序列。
预期每个单独序列的pcr扩增根据扩增子的长度产生序列亚结构域的可变的产量,因此将序列分成多个重叠子序列,各1200bp。所有亚片段被设计为包括与相邻亚片段至少100bp重叠以适应引物位置的变化。该处理产生95个候选序列。
对于每个序列,使用基于网络的程序“batchprimer3”设计引物对,所述程序寻找通用引物以产生这样的扩增子:最小800,最佳1200,最大1200;引物长度最小22,最佳25,最大30;tm最小65,最佳70,最大75。引物序列作为xls文件下载并复制到电子表格中以调整格式使其适合放置订单。
引物从idt订购为混合的正向和反向引物12nmole,各自在深孔板中干燥的。向每个孔中加入240μl水(5prime2900132)以得到各自50um。根据pcr需要制备稀释液。
基因组dna的pcr对于仅约一半的孔产生干净的1200聚体产物,因此使用含有期望的基因座的bacdna的制备物来产生高得多的成功率。覆盖该基因座的大肠杆菌中的bac克隆从genomesystems,inc.st.louis获得,进行培养物生长并通过“mini-prep”分离dna。
pcr主混合物:使用含有phirehotstartiidna聚合酶(thermof-122l)的pcr试剂盒。它含有400μl5x反应缓冲液+40μl25mmdntp(rochediagnostics,indianapolis,in)+40μlphire聚合酶+60μldmso+1260μl水+0.5ugbacdna模板。
pcr:向96孔板的孔中加入2μl每个引物对5um+18μl主混合物;将孔盖上盖,放在热循环仪上,程序为98度30秒,30x(98度8秒,72度30秒,72度2分钟),72度10分钟,4度。在完成后,将5μl的每个反应物取样到具有痕量的6xdna上样染料(thermor0611)的20μl水中,将20μl转移到egel96琼脂糖凝胶(lifetechnologies)的孔中,并电泳8分钟。96个孔中的94个在期望的mw显示干净的条带(图3)。
扩大pcr以将产量最大化。向含有来自电泳样品的剩余5μl的每个孔中,加入10μl相同的5um引物+10μl水+25μl的dreamtaqgreen2xpcrmm,并将管盖上盖并放置在具有程序10x(95度30秒,55度30秒,72度2分钟)72度6分钟的热循环仪上。完成后,向每个孔加入6μl的(30μl1mmgcl2+192μl25mmdntp+378μl水+100μldreamtaqgreen2xpcrmm)并将管盖上盖并放置在具有程序6x(95度30秒,55度30秒,72度2分钟)72度10分钟的热循环仪上。
将柱的内容物合并以得到表示基因组范围不同基因座的6个扩增子混合物,并通过用异丙醇沉淀分离dna,并重悬于水中,得到含有52-75μgdna的100μl溶液。
通过超声处理片段化:将来自pcr孔1-48和49-96的约180μgdna1200聚体在管中与水合并以得到400μl,且乙酸钠为300mm。将管放置在超声波仪柜内的冰水烧杯中,超声处理(超声波仪branson450)输出控制3,30%占空比16分钟。超声处理产物用异丙醇沉淀,并重悬于200μl的20mmtrisph8.2中的600mmnacl中。通过以下将样品分级:具有monoq柱的hplc(gelifesciences),缓冲液a=20mmtrisph8.2;缓冲液b=a+2.0mnacl,0.4ml/min%b=40-50经32分钟,收集级分。dna洗脱在宽峰中,中心为约15分钟。合并级分得到各自7个合并物。用溴化乙锭显现的3%琼脂糖凝胶上的电泳显示范围为约80bp至约400bp的级分(标记的h15、h18、h21、h24、h27、h30、h33)(图4)。
片段化的dna的胺化:合并的级分通过异丙醇沉淀浓缩,重悬于20μl水中并通过在沸水中加热1分钟来变性。向20μl变性的dna中加入180μl的1000μl水、600μl三氟乙酸(sigma,stlouismo)348μl乙二胺和190mg焦亚硫酸钠的混合物,并将混合物在65℃下孵育20分钟。然后将混合物通过sephadexg25脱盐,并将脱盐产物通过异丙醇沉淀浓缩并再悬浮于水中。
通过本领域熟知的方法,用羧基四甲基罗丹明使用其nhs酯(lifetechnologiesc1171)将胺化的产物标记。通过使用10kda滤器(nanosep10omega,pallcorporation,annarbor,mi)的超滤从剩余的未结合染料中分离标记的产物,然后经0.22u过滤器(milliporeufc30gv00,billerica,ma)过滤。
经衔接子介导的pcr制备探针
还通过连接衔接子并使用产物作为pcr模板使用对应于衔接子序列的引物修饰所需大小的片段。这提供了产生更大量的期望大小的产物的有效方法,以及提供将额外官能团偶联到产物上的方法。该方法由以下组成:修饰片段以产生5'磷酸化的平末端,然后连接期望序列的衔接子。衔接子可以被设计成含有限制位点,使得dsdnapcr产物可以用合适的限制性酶切割以除去不想要的衔接子序列或以露出适于连接另外的基团的粘性末端。
通过使用thermofastdnaendrepairkitk0771根据说明书进行片段的末端修复,用4ug各级分h18、h24、h30开始。使用离心柱(invitrogenk310001)分离平端产物。处理4ug超声处理的分级的dna产生约3.5ug平端产物h18b、h24b、h30b,各自的浓度为74、66、70ng/μl。
通过将每种寡核苷酸g6a、g6b、gc6a、gc6b、bspd、bstb、bspdc、bstbc与20mmtrisph8.0和250mmnacl组合来制备衔接子混合物,将混合物在沸水中加热1分钟并冷却至室温。t4dna连接酶(invitrogena13726,含有以5单位/μl的连接酶和反应缓冲液的试剂盒)用于将衔接子连接到各平端的dna级分:向10μl平端的dna级分中加入5μl衔接子混合物、2μl10x连接酶缓冲液、2μl50%peg4000和1μl,并将混合物在室温下放置过夜。使用旋转柱(invitrogenk310001)在50μl洗脱缓冲液中洗脱来分离衔接产物,且标记的h18t、h24t、h30t各自浓度为37、39、30ng/μl。
衔接的her2级分的pcr以20μl规模各自使用1μl的衔接模板h18t、h25t、h30t和商购pcrmastermix(dreamtaqgreenthermok1081)进行。各反应物含有5或10um的单一引物g6a或bspd。循环条件为24x(95度30秒,52度30秒,72度30秒)。通过向4μl产物中加入1μl1m氢氧化钠使产物变性,并在3%琼脂糖/etbr凝胶上分析。对于所有测试的引物和引物浓度,pcr产物显示对应于模板大小的条带(图5)。
使用图5所示的pcr的产物作为使用10umbspd24引物扩增的模板;加入10μl的bspd10-18、bspd10-24和bspd5-30以得到在最终的1x主混合物中的800μl。将其分成8×100μl份,并用循环条件12x(95度30秒,60度30秒,72度2分钟)扩增。向每个孔中加入额外体积2.1μl的41.5μl25mmdntp(rochediagnostics,indianapolis,in)和13μl1.0m氯化镁(sigma,stlouismo)的混合物,并将混合物进行额外的8x(95度30秒,60度30秒,72度4分钟)。将合并的产物用异丙醇沉淀,各自再悬浮于200μl水中,并用聚乙二醇进一步沉淀以从引物分离pcr产物。将最终pcr产物溶于200μl水中,标记h18b、h24b、h30b,浓度测量为1126、1270、1357ng/μl。
pcr产物的限制性消化以减少衔接子部分:使用限制酶bspdi(newenglandbiolabsipswichma)。向260μl水中加入40μl10scutsmart缓冲液bspdi(newenglandbiolabsipswichma)、100μl上述pcr产物h18b、h24b、h30b和10μl10u/μlbspdi,并将混合物在37℃下孵育16小时。将反应物标记为h18r、h24r、h30r。
16小时孵育后,取各样品用于电泳,比较消化与未消化的产物。向pcr条的孔中加入8μl2x上样染料(thermofisherscientific)、0.6μl(h18b、h24b、h30b)、2.4μl(h18r、h24r、h30r)+水至10μl;各混合物分为各自2x5μl,所有中均加入4μl水。向每一个中加入1μl1mnaoh并加热95度30秒以变性。各自5µl上样到3%琼脂糖/etbr凝胶的孔中并电泳。在凝胶图像中,“-”和“+”对应于不存在和存在naoh。所有消化产物都显示对于bspdi成功切割所预期的较小大小和末端片段的存在(可能来自与残留引物的退火片段的双体(doublet))。naoh的变性显示对未消化和消化的产物两者大小的进一步降低。通过变性消除来自片段延长退火的较高分子量的弥散(smear)。
产物h18b、h24b、h30b、h18r、h24r、h30r用异丙醇沉淀,再悬浮于20μl水中,并通过上述程序进行胺化和用羧基四甲基罗丹明标记。
fish杂交条件
靶向探针与结合玻璃显微镜载玻片的淋巴细胞中的人染色体dna杂交。在典型的实验中,试剂混合物由以下组成:7μl的lsi/wcp杂交缓冲液(abbottmolecular(desplaines,il))和3μl含有2000ng超声处理的人胎盘dna、500ngcot-1dna(lifetechnologiestm(grandisland,ny))、50ng的探针cep17-sg缓冲液(abbottmolecular(desplaines,il))和100ng的测试探针的水。
将显微镜载玻片通过连续浸入70%、85%和100%乙醇中脱水,然后风干。将测试溶液(10μl)放置在载玻片上并用22×22mm的盖玻片覆盖,使溶液铺展在覆盖区域上。施加橡胶胶水以将边缘密封并将载玻片放置在控制温度的仪器中。将温度升至70℃持续5分钟以使样品和试剂的dna变性,然后降至45℃1小时,以允许试剂与其靶标杂交的时间。杂交时间完成后,去除橡胶胶水和盖玻片,将载玻片在0.4×ssc和0.3%np40的溶液中在73℃下洗涤2分钟,然后在2×ssc、0.1%np40中在室温下洗涤1分钟,然后风干。
通过将10μl的dapi溶液置于目标区域上并用盖玻片覆盖来制备载玻片用于观察。用装有适合于目标荧光团的滤光片的荧光显微镜观察载玻片。
所得的杂交图案的荧光显微术
将10μl的dapi-ii(abbottmolecular(desplaines,il))置于载玻片上靶标位置处,用22×22mm盖玻片覆盖,并在配备有允许同时显现dapi、荧光素(绿色)和tamra(橙色)信号的滤光片的荧光显微镜下观察。照片(图7)显示橙色和绿色信号的图案与对于由测试探针染色的her2基因座(橙色)和由cep17-sg染色的着丝粒位置(绿色)所预期的那些相符。在中期染色体上,橙色和绿色信号位于同一染色体上的相邻位置,而间期核在任意距离对各自显示两个强信号。对于经相同hplc级分的衔接子介导的pcr制备的探针可以看到类似的结果。在标记之前通过限制性消化去除衔接子末端产生具有无法区分的性能特征的探针。
用于1200聚体片段的引物序列
衔接子修饰和衔接子介导的pcr中所用的序列
实施例2
基于用于比较的可获得的bac探针,使用模型靶标p53、her2和p16进行设计。用ucsc基因组浏览器鉴定与靶标相对应且标记了重复序列用于去除的基因组序列。
对于pcr探针,使用计算机应用来分离序列的无重复片段部分,并鉴定针对靶标的片段用于制备为“千聚体(kilomers)”。使用基于网络的应用“batchprimer3”产生引物序列以扩增尽可能多的每个千聚体序列,并且通过idt以96孔格式合成鉴定的引物序列。对于寡聚物和寡聚物-pcr杂交探针,进一步处理序列的无重复片段部分以鉴定指定大小和gc含量的片段。该处理使用excel电子表格来附加通用衔接子序列、分选和将序列列表格式化用于放置合成订单。
通过寡聚物合成、寡聚物-pcr杂交或通过1步或2步pcr产生以最终探针结束的大量dna。1步pcr方法如下:在使用bac或基因组dna模板的pcr后,将产物合并并通过超声处理片段化以得到可通过与基于bac的fish探针使用的相同的方法进行化学标记的产物。这样制备的fish探针在结构上与ambac探针相同—唯一的区别是pcr探针排除了bacdna中存在的重复序列和载体序列。2步方法如下:将超声处理的产物与衔接子连接以制备含有所有选择的靶序列的单一模板混合物。该模板然后可用于使用单一组引物的单一pcr反应以产生用于胺化和标记的大量dna。在这种情况下,超声处理不再需要,因为扩增产物已经是期望的大小。将模板制备一次,无限期存储并为每个新制剂采集样品。衔接子序列存在于产物的5'和3'末端。虽然它们可以通过限制性消化来去除,但测试显示它们的存在不会损害fish测定中的性能。
通过在pcr反应中包含氨基烯丙基dutp或通过化学胺化引入用于连接荧光团标记的胺基。化学胺化通过使用am探针的亚硫酸氢盐/tfa/乙二胺方法进行,但例外是在将反应混合物脱盐之后但在乙醇沉淀之前加入少量的四甲基乙二胺。这替代残留的未连接的乙二胺,否则乙二胺会在标记反应中竞争荧光团。
用荧光团标记胺化的dna是通过更适合于许多小规模反应的已建立方法的改变来完成。在该改变中,将胺化的dna与四甲基乙二胺和氯化钠在25%dmso中的反应缓冲液合并,加入活性荧光团,并将混合物在60℃下孵育2小时。通过乙醇沉淀分离产物,并进行75℃72小时甲酰胺处理标准。
her2pcr探针:
对于her2pcr探针,产生三种变体,均使用76kb的无重复片段的序列。对于最简单的“1步pcr”探针,将pcr产生的dna与常规探针中的bacdna相同处理:通过超声处理片段化,然后进行化学胺化和标记。对于“2步pcr”探针,将超声处理片段连接到衔接子以制备模板。用单一引物扩增该模板以产生准备进行胺化和标记的大量dna。在第三种形式中,“用氨基烯丙基dutp的2步pcr”,在氨基烯丙基dutp存在下用引物对扩增该相同的模板以生成胺化的产物,准备用于用任何期望的荧光团进行标记。
her2基因座中无重复片段序列的产生:
使用通过p1克隆pvys174c,e,h,i限定的基因座鉴定her2pcr形式的序列。这些克隆包括在hg18_dna范围chr17:35004678-35230380的226kb。这些坐标被输入到ucsd基因组浏览器中,并且使用repeatmasker功能呈现相应的序列以将已知为重复序列的部分呈现为小写字母。使用应用复制大写字母的“唯一”序列部分,以选出大写字母唯一子序列,保留各自的位置信息。这产生了长于1200bp(1234至5615bp)的35个序列,总计为76904bp。用另一个应用处理这些序列,将所有序列分成总共95个序列,每个序列1200bp,且具有至少300bp重叠。将1200bp序列输入基于网络的应用“batchprimer3”,调整设置以产生具有至少65度的tm的引物,以在扩增子中包含尽可能多的(至少800bp)的每个序列。
合成鉴定的引物序列并将其作为引物对置于96孔板形式中。将引物溶解在水中并制备以得到各5um的引物,同时仍在96孔板形式中。pcr以如引物对的相同形式在96孔板中使用含有phire聚合酶、由p1克隆pvys174c,e,h,i的混合物组成的模板和以0.5um的引物的主混合物进行。(注意,可以使用基因组dna作为模板,但优选基因座特异性克隆诸如bac和pac的)。通过96孔egel分析产物,并在95个孔的94个显示强且干净(strongclean)的条带。当使用taq聚合酶时,95个反应中仅87个显示产物。
her-2无重复片段序列的片段化:
合并pcr孔的内含物并通过乙醇沉淀和peg沉淀分离dna1200聚体。将1200聚体混合物通过用于将其他am探针的bacdna片段化的相同方法进行超声处理,并将超声处理的产物通过具有离子交换柱的hplc分级以得到范围在约120-约400bp的窄大小级分。在一些实施方式中,取消分级步骤。
来自1步pcr方法的her-2探针:
为了制备探针5/13-76a,将中心位于150bp的级分通过标准程序胺化并用spectrumorange标记。
来自2步pcr方法的her-2探针:
为了从两步法制备her-2探针,将一部分相同的150bphplc级分用平端化剂处理并与衔接子连接。在第二次pcr反应中使用衔接产物作为模板,这次仅使用单一引物,其序列对应于衔接子。使用单一引物扩增衔接模板通过茎-环结构抑制最短片段的扩增,并产生富集较长片段的产物。将pcr产物胺化并用spectrumorange标记,然后进行75℃72小时甲酰胺处理。
经氨基烯丙基dutp胺化的her-2pcr探针:
为了通过氨基烯丙基dutp胺化制备her-2pcr探针,将超声处理的1200聚体的hplc级分重组(以模拟未分级片段),用平端化剂处理并连接到包含含有bspqi限制位点的通用5'末端的衔接子上。在第二次pcr反应中使用衔接产物作为模板,这次使用对应于衔接子序列的两个引物,并且用氨基烯丙基dutp替代一半的dttp。用限制性酶bspqi处理pcr产物以消化共同的末端,并用spectrumorange标记的dna产物,然后进行75℃72小时甲酰胺处理。
her-2寡聚物探针:
设计her-2寡聚物探针开始于如her2pcr探针的相同无重复片段序列。使用应用来选择该序列的无重叠80个碱基的部分,均具有大约50%gc含量(g+c最小=39,最大=41)。发现总计401个此类序列。将这些输入网络应用quickfold,参数为na+=0.05m,mg++=0.001m,最大1折叠(folding)且结果复制为文本和δg。使用excel的发现特色来标记含有去除某些限制位点的序列(以允许这些序列用于衔接子而不干扰靶序列)。其余序列通过δg分选,并选择具有最低折叠倾向的288(三个96孔板)用于进一步处理和通过基因组定位分选。80聚体序列通过通用20个碱基连接序列成对连接,并且还将不同的通用20个碱基加入到该对的3'端,使总共待合成200个碱基。全部288个80聚体(图代表144个单独的“正向”200聚体。还计算了靶标80聚体和通用20聚体的互补体,并以相同的方式装配以提供144个单独的“反向”200聚体。这些装配被设计成使得在混合时,序列应以交错的方式形成双链体,产生退火产物的长链。
核酸通过上述程序进行化学胺化,并用spectrumorange标记。通过凝胶电泳纯化标记产物以除去截短的合成产物,并将全长产物组合以得到最终探针。
额外的her2寡聚物探针的设计开始于与her2pcr探针的相同序列,掩蔽重复序列。引入额外的掩蔽来标记去除其他不需要的子序列:5个或更多个连续的“g”或“c”碱基,以及与bspqi限制位点“gctcttc”和“gaagagc”相对应的序列。处理剩余的序列以鉴定具有55-65%gc的60个碱基的段。在序列下游40kb中发现非常少的可接受的序列,因此只有1-180kb范围内的序列进入到下一阶段。使用互补引物向每个序列的3'末端加入碱基“ggttgaagag”聚合酶。使用基于网络的应用“zipfold”来测定折叠的能量。在评估的1027个60聚体中,保留具有最低折叠倾向的960个。根据折叠能量将它们分成5组(每组192个成员=两个96孔板),每组按整个基因组序列中的位置分选。将寡核苷酸溶解在水中,并根据折叠趋势组合成组。每个组的样品用含有对应于通用3'末端序列的引物和phire聚合酶进行退火,以延伸各自至平端双链体混合物。这些通过离子交换hplc纯化以除去截短的寡聚物的产物。
p53pcr探针:
与her-2探针相同,基于相应的bacp53探针的bac的序列设计了p53pcr探针。千聚体通过pcr产生,合并并通过超声处理片段化。对于1步pcr探针,将超声处理片段简单地胺化并标记。对于2步pcr变体,将超声处理片段平端化并与衔接子连接以制备模板。为了产生用于胺化和标记的大量dna,使用对应于衔接子序列的引物扩增模板。从相同模板(但在反应混合物中包含氨基烯丙基dutp)制备含有胺用于标记的大量dna。对于另外的变体,通过用对于包含在衔接子中的识别位点特异性的限制酶消化pcr产物来除去衔接子序列。
p53基因座中无重复片段序列的产生:
使用通过bac克隆pvys173i限定的基因座鉴定p53pcr形式的序列。该克隆由在hg18_dna范围chr17:7435119-7606823的172kb组成。这些坐标被输入到ucsd基因组浏览器中,并且使用repeatmasker功能呈现相应的序列以将已知为重复序列的部分呈现为小写字母。下表1显示了引物序列。
如上所述复制大写字母的“唯一”序列部分,以选出大写字母“唯一”子序列,保留各自的位置信息。这产生长于1200bp的仅17个序列,总计35930bp。向这些中加入13个具有长度800-1200的片段和10个650-800bp的片段以得到最终总计55661bp。对第一组使用应用“seqchop3”以产生具有400个碱基重叠的1200聚体,得到65个序列。具有200个碱基重叠的800聚体的第二组(800-1200)产生26个序列。为了完成96孔板,接受650-800碱基区域中的额外5个序列。调整这样产生的序列以得到tm至少65度的引物,以在扩增子中包括尽可能多的每个序列。将引物溶解在水中以得到各5um的引物,同时仍在96孔板形式中。pcr以如引物对的相同形式在96孔板中使用含有phire聚合酶、由bac克隆pvys173i组成的模板和以0.5um的引物的主混合物进行。pcr完结时,加入额外引物、taq聚合酶和dntp,并将板进行额外8个循环以增加产物产率。通过96孔egel分析产物,并在96个孔的91个显示强且干净(strongclean)的条带。合并pcr孔的内含物并通过乙醇沉淀和peg沉淀分离dna。通过用于将其他探针的bacdna片段化的相同方法将混合物超声处理。
来自1步pcr方法的p53探针:
为了从1步pcr方法制备p53探针,将超声处理的产物胺化并用spectrumorange标记,随后进行75℃72h甲酰胺处理。
来自2步pcr方法的p53探针:
为了从2步pcr方法制备p53探针,将超声处理的产物的一部分使用一次性硅胶基离心柱(purelinkpcrpurificationkit)分级,但调整结合缓冲液以分离范围在100-300bp的级分。该级分的一部分用平端化剂处理并连接至衔接子。在第二次pcr反应中使用该衔接产物作为模板,这次仅使用单一引物,其序列对应于衔接子。将pcr产物胺化,用spectrumorange标记,并进行75℃72小时甲酰胺处理。
经氨基烯丙基dutp胺化的p53pcr探针:
如上所述制备该探针,例外是pcr步骤包括加入到pcr混合物中的氨基烯丙基dutp。将pcr产物用spectrumorange标记,并进行75℃72小时甲酰胺处理。
将一部分超声处理的平端化的无重复片段的p53dna连接至含有bspqi限制位点的衔接子。将产物用作在含有氨基烯丙基dutp的pcr反应(具有一对对应于衔接子的引物)中的模板,并将产物暴露于bspqi限制酶以去除衔接子末端。仅含有p53特异性序列的消化产物用spectrumorange标记,然后进行7℃72小时甲酰胺处理。
p53的寡聚物pcr探针
p53寡核苷酸探针的序列如上鉴定;然而,在这种情况下,除了掩蔽重复序列之外,掩蔽了5个或更多个连续的“g”和“c”的所有子序列。设置参数以鉴定长度为140且gc含量为45-60%的子序列。将鉴定的289个序列进行ncbiblast以找到与人基因组中其他基因座的匹配。此后仍有281个唯一序列。向这些中的280个附加了各25个碱基并含有限制位点的通用正向和反向序列以得到各190个碱基的280个序列。将这些序列送到idt用于以96孔板格式合成为“超聚体(μltramers)”。将产物合并,并在pcr中用作用对应于衔接子的引物序列的模板。通过用限制酶消化去除衔接子末端,并将产物胺化,用spectrumorange标记,并进行75℃72小时甲酰胺处理。
表2显示了190bp序列。
表3显示探针设计和下游/清除(cleanup)方法的概述。
表3
测试结果
探针首先在雄性淋巴细胞载玻片上测试,并且评价探针密度、特异性、背景和交叉杂交,用对应的bac探针作为对照。
所有测试的探针通过了在淋巴细胞载玻片上的交叉杂交测试。一旦探针设计在淋巴细胞载玻片上通过,则随后将其在ffpe载玻片上测试。所有探针在乳腺癌组织ffpe载玻片上测试。一些探针在乳腺和胃部载玻片上测试。对于所有ffpe载玻片使用通用的预处理方法。使用来自am库存的her2和p53bac探针作为所有实验的对照。bac对照探针通过了在淋巴细胞载玻片和ffpe组织载玻片上的过夜杂交,但没有通过在两种载玻片类型上的1小时杂交。
结果表明rfpcr探针设计优于通过直接合成或寡聚物pcr杂交物设计的合成寡聚物的性能。直接合成的her2寡聚物探针和寡聚物pcr杂交物p53探针没有通过1小时ffpe,而寡聚物pcr杂交物her2探针没有通过所有质量评价。基于这些数据,将寡聚物探针设计从进一步评价中排除,而rfpcr方法用于选择最佳探针设计。
所有rfpcr设计均通过了在淋巴细胞和ffpe载玻片上的1小时和过夜杂交的质量评价,除了在淋巴细胞载玻片上观察到的一些次优特异性数据点。
2步pcr设计具有若干下游或清除选项,诸如超声处理后的dna片段分级、用氨基烯丙基dutp的第二pcr和第二pcr后的衔接子去除。表1列出了每一探针设计的所有下游或清除步骤。对于两种her22步探针,不同的分级大小、第二pcr具有或不具有氨基烯丙基dutp和第二pcr后去除或不去除衔接子对于最终探针质量没有正面影响。
与三种p532步pcr探针相同,第二pcr具有氨基烯丙基dutp相对于胺化和第二pcr后去除或不去除衔接子不影响最终探针质量。
无重复片段的pcrfish探针设计和制备的最终程序如下。
1:在基因组浏览器上鉴定期望的基因座坐标。
2:下载具有重复片段掩蔽为n的序列。
3:将序列输入序列处理程序中。
4:处理以鉴定千聚体的唯一序列,并对batchprimer3格式化。
5:使用batchprimer3(网络)以鉴定各千聚体的f和r引物。
6:订购以96孔板形式的混合的引物。
7:使用phusion聚合酶在板中pcr,通过egel-96评估。
8:合并pcr产物,通过超声处理片段化。
9:将超声处理的dna平端化。
10:连接衔接子以制备模板。
11:用以下pcr:衔接子引物以制备大量用于胺化、标记的dna。
或衔接子引物和aadutp以制备用于标记的胺化的dna。
13:(如需要)胺化
14:标记,甲酰胺处理。
实施例3
该实施例描述了靶向p16的无重复片段的探针的开发。p16探针的图谱显示在图9中。引物序列显示于表4中。
表4
1200聚体用phusion的pcr:向2µl各p16rf5um加入18µlphusionmm并置于热循环仪,程序为98度20秒,25x(98℃8秒,68℃30秒,72度1分钟),72℃10分钟,4℃。延伸pcr:向各孔加入2µl的相同5µm引物和2µl(80μl2xpcrmm+60µldntp+20µl1mmgcl2)并进行热循环:10x(98℃8秒,68℃30秒,72℃2分钟)。将3µl上样至egel的孔,电泳8分钟。大部分孔(68/73)显示出预期(相同)mw处的强且干净的条带。然而,一些显示紧密靠近的双带(doublet)。将p16rfphusionpcr的孔合并至总共1200μl,在管中分成3×400μl,并加入40μl3mnaac和1mletoh,并将管置于-20℃。将管以15000rpm4℃离心12分钟,在400μl70%etoh中洗涤沉淀,真空离心机干燥,合并,再悬浮于200μl水中,置于65℃烘箱中5分钟,以4000rpm离心5min沉淀聚合酶。将上清液转移至新管,加入300µlpegrgt。15分钟后,将管以14000rpm离心5分钟,将沉淀重悬于400µl水。电泳显示条带与1200聚体混合物一致。
将p16-1200聚体超声处理:(保留20µl用作模板)向380μlp16-1200聚体加入40μl3mnaac,将管以3,30%占空比2x10分钟超声处理,并加入1mletoh。将管以15000rpm4℃离心10分钟,将沉淀在400µl70%etoh中洗涤,干燥,并重悬于100µl水中。将管随后以15000rpm4℃离心5分钟以沉淀来自超声仪探头的残留物。电泳显示大约50-400bp的大小范围。
分级:向10μl超声处理的1200聚体中加入40μl水和10μl缓冲液,将管混合并吸取至离心柱1,14000rpm离心1分钟;向流出液加入10μl缓冲液,混合,吸取至离心柱2,14000rpm离心1分钟;向流出液加入10μl缓冲液中,混合,吸取至离心柱3,14000rpm离心1分钟。将柱用2x200µl洗涤缓冲液洗涤并用50µl洗脱缓冲液洗脱。电泳显示大约100-300bp的大小。
使用fastdnaendrepairkit将p16son和p16s2平端化:向于40µl水中的3µl的上述物质加入5µl的10x缓冲液2.5μl的酶混合物并将管涡旋并置于20℃水浴持续20分钟。根据说明书使用inversepcr纯化试剂盒(lifetechnologies)来分离产物于50µl缓冲液中。
连接衔接子至p16sonb,p16s2b:将1µl各1000µmpadbspd1、cadbspd1、padecor1、cadecor1、5µl10x连接酶缓冲液、5µl50%peg4000、5µl7/28(p16sonb,p16s2b)加入各管中,将管涡旋并加入1µl连接酶。使用inversepcr纯化试剂盒来分离产物。
衔接的p16的pcr:向条的孔中加入(8,8,2,8,2)µl水和(1µl各20µm,2,8µl20umpadbspd1;2,8µl20umpadecor1)。向孔中加入10µl的50µldreamtaqgreen2xpcrmm+2µl的以上探针。热循环用程序25x(95℃30秒,55℃30秒,72℃30秒)进行。分析显示来自p16sona模板的产物条带比来自p16p2a的产物条带稍微更宽。对于两者,2µm的单一引物得到类似于8µm的产量;padbspd1和padecor1引物之间没有差别。padecor1产生最佳的总体性能。
用padecor1单一引物5µm的衔接p16的制备(prep)pcr。向条的孔中加入100µl(400µl水+4µl1000µmpadecor1+2µl(p16sona,p16s2a)+200µldreamtaq2xpcrmm),加盖,置于热循环仪上,程序为25x(95℃30秒,55℃30秒,72℃30秒)。随后,向每孔中加入额外3.0µl(30µl25mmdntp+10µl1mmgcl2+8µl1000µmpadecor1)并用以下程序进行热循环:10x(94℃30秒,55℃30秒,72℃2分钟)。将产物沉淀并如上重悬。
将p16制备物胺化:转移50µl以上p16son至管中并用真空离心机降低体积至20µl。将其和剩余20µl7/29p16sp,p16sp2p置于沸水浴中1分钟,向各自中加入180µl(500µl水+300µltfa+174µl乙二胺+95mgna2s2o3并置于65℃水浴持续15分钟。随后,将溶液脱盐入水中并收集450µl的各纳滴(nanodrop)。加入tmed/nacl,将管置于65℃浴中5分钟,加入50µl3mnaac和1ml异丙醇。将所得溶液沉淀并重悬于水中。
用tamra,cr6g标记胺化的p16产物:向上述中加入1µl1mnaoh;1分钟后,加入25µl(75µltmed/nacl+75µldmso),将管涡旋并加入2µl(100mmtamra,100mmcr6g)。将所得溶液沉淀并重悬于水+15µl20xssc和105µl甲酰胺以得到2xssc中的150µl。所得溶液如上纯化。
本申请提及和/或下文列出的所有公开和专利均通过引用并入本文。所述的特征和实施方案的各种改变、重组和变化对于本领域技术人员是显而易见的,且不会背离本发明的范围和精神。尽管已经描述了具体的实施方案,但应理解要求保护的本发明不应过度地限于此类具体的实施方案。实际上,对于相关领域中的技术人员显而易见的所述方式和实施方案的各种改变均旨在以下权利要求书的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!