大环内酯类衍生物及其用途的制作方法
本发明涉及药物化学技术领域,进一步涉及新的化合物及其用途,具体涉及大环内酯类衍生物及其用途。
背景技术:
消化道功能性疾病是临床上最常见的疾病之一,其中绝大多数病人存在消化道运动障碍,即消化道动力不足。正常健康人的结肠运动包括短时相性收缩、长时相性收缩、张力性收缩和巨大移行性收缩(GMCs)。一般情况下结肠多处于静止或低幅非推进性时相性收缩状况,而GMSs每天出现1-2次,引起集团运动,与排便有关,胃结肠反射是诱发结肠运动的一种主要形式。绝大多数消化道功能障碍病人无特异性疾病。功能性便秘病人主要分为结肠通过时间延缓型和通过时间正常型两类,前者病人可出现GMCs频率、持续时间及幅度降低、胃结肠反射减弱或消失,而局部时相性非推进性收缩增强,使得整个结肠运动不协调,少数病人出现小肠运动减弱。通过时间正常的患者主要存在肛门括约肌功能异常和直肠感觉功能下降。这些组织结构的功能依赖于各种神经递质和体液因子的释放而建立联系,各种递质和信使与相应的受体结合,执行不同的生理功能。因消化道动力不足引发的消化不良、胃胀、胃食管反流、动力性肠梗阻、便秘、大便干燥、肠蠕动乏力,肠易激综合征等疾病严重影响患者的日常生活。
目前的促消化道动力药物的靶点选择性较差、副作用较多,因此,开发一种靶点选择性强、副作用较少的促胃肠动力药物具有深远的临床意义和广阔的市场前景。
大环内酯类化合物普遍具有抗生素活性,长期服用具有抗生素活性的药物,会引起体内菌群的耐药性,副作用大。大环内酯类衍生物作为其他医药用途时,其抗生素活性所带来的副作用大大限制了药物的开发和使用。
技术实现要素:
本发明旨在针对现有技术的技术缺陷,提供一种大环内酯类衍生物及其用途,以解决现有技术中缺乏一种类似化合物的技术问题。
本发明要解决的另一技术问题是现有技术的促胃肠动力药物疗效有待提升。
本发明要解决的再一技术问题是现有技术的大环内酯类化合物或其衍生物因具有抗生素活性、易导致副作用的问题而限制了其他制药用途的技术问题。
为实现以上技术目的,本发明采用以下技术方案:
大环内酯类衍生物,其特征在于具有如式(1)所示的结构:
其中,R1选自氢、羟基、卤素、C1-C6烷基、C1-C4烷氧基、C1-C6链烯基、(CH2)m(C6-C10元芳基)、(CH2)m(C5-C10元杂芳基)或C2-C10炔基;R2、R3均选自氢、羟基、卤素、C1-C6烷基、C1-C6链烯基、(CH2)m(C6-C10元芳基)、(CH2)m(C5-C10元杂芳基)、C2-C10炔基、C1-C4烷氧基、吡喃糖基,其中,所述吡喃糖基是未取代的或者被下列基团中的一个或多个取代:氢、卤素、C1-C6烷基、C1-C4烷氧基、C1-C4烷酰基、C1-C4烷酰氧基;每个m独立地为0-4的整数。
作为优选,所述大环内酯类衍生物选自化合物III-1、化合物III-2、化合物III-3、化合物III-4、化合物III-5,这些化合物的结构如下所示:
上述大环内酯类衍生物在制备促进消化道动力药物方面的用途。
上述大环内酯类衍生物在制备辅助促进消化道动力药物方面的用途。
作为优选,所述促进消化道动力药物的适应症为消化不良、胃胀、胃食管反流、肠蠕动乏力,动力性肠梗阻、便秘、大便干燥、肠易激综合征等引发胃肠动力不足的一种或多种疾病类型。
作为优选,所述促进消化道动力药物包括:
化合物III-1、化合物III-2、化合物III-3、化合物III-4、化合物III-5或它们的组合;或者化合物III-1、化合物III-2、化合物III-3、化合物III-4、化合物III-5在药学上可接受的酸、碱、盐、酯、水合物或它们的组合。
作为优选,所述促进消化道动力药物的剂型选自注射剂、片剂、胶囊剂、溶液剂、颗粒剂、滴剂、散剂、糖浆剂、酒剂、酊剂、露剂、膜剂、丸剂、气雾剂、软膏剂、栓剂、洗剂或它们的组合。
作为优选,所述促进消化道动力药物的给药方式包括:口服、注射、植入、外用、喷雾、吸入或它们的组合。
本发明提供了一种全新结构的大环内酯类衍生物,并通过实验手段发现其具有很好的促进消化道动力的作用,能增强肠蠕动能力,增加排便量,可加快肠内容物通过;在此基础上,本发明进一步发现上述大环内酯类衍生物抗生素活性低、副作用小,可作为促胃肠动力药服用。特别的本发明还对筛选出的药效最好的化合物III-3,利用家兔,比格犬,狨猴进行了进一步的药效评价,并评价了其急性毒性和心脏毒性,实验结果表明化合物III-3具有很好的促进胃肠道动力的作用及安全性,具有很好的成药性。
附图说明
图1示出了大环内酯类衍生物的合成路线图。
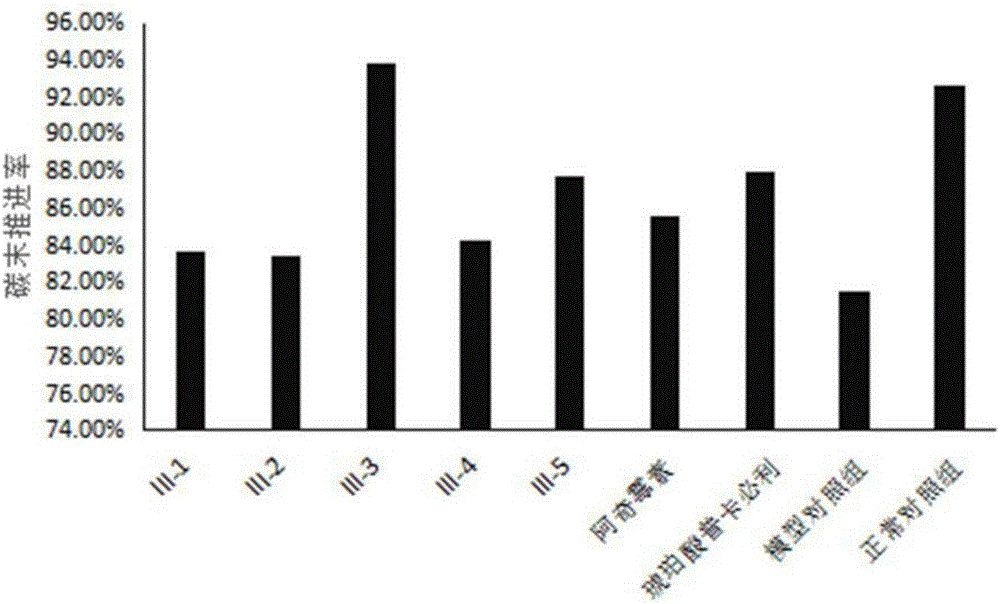
图2大环内酯类衍生物对肠蠕动乏力模型的影响结果图。
图3示出了化合物III-3对兔子、比格犬、猴子的排便重量及排便粒数的影响。
图4示出了化合物III-3对大鼠的心脏毒性检测结果。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。以下所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
本发明通过化学反应制备了化合物大环内酯类衍生物,通过1H-NMR(核磁共振氢谱)和MS(质谱)对大环内酯类衍生物进行了结构鉴定。通过一系列实验检测大环内酯类衍生物的抑菌活性和促进肠道动力的活性。具体实施方式如下:
下列实施例中,所用的试验材料及其来源包括:
(1)小鼠
昆明小鼠(雌性):由中国人民解放军军事医学科学院试验动物中心和北京维通利华实验动物技术有限公司提供。
动物到达后,由专人接收动物于双走廊屏障环境小鼠饲养室内,填写《试验动物接收记录表》(BG-017-V00),接收时对动物大体情况进行观察,并随机抽取动物进行称重,确保试验动物与引进标准基本吻合。实验动物使用许可证号:SYXK(津)2012-0003。
(2)供试品
阿奇霉素:白色或类白色结晶性粉末,购自大连美仑生物技术有限公司,批号:20130216,纯度:94.5%(符合中国药典2010年版二部)。
硫酸阿托品:白色或类白色结晶性粉末,购自大连美仑生物技术有限公司,批号:20130824,纯度:98.5%(符合中国药典2010年版二部)。
琥珀酸普卡必利:白色或类白色结晶性粉末,购自上海法默生物科技有限公司,批号:20131010,纯度:99.45%(符合中国药典2010年版二部)。
盐酸购自利安隆博华(天津)医药有限公司;氢氧化钠购自天津市化学试剂供销公司;无水硫酸镁购自天津市光复精细化工研究所;二氯甲烷购自天津市化学试剂供销公司;大肠杆菌来自天津国际医药联合研究高通量药物筛选中心。LB培养基中用的牛肉膏和蛋白胨,都购自OXOID公司。
供试品于室温保存。
(5)所用药物及试剂的配制方法包括:
a)阿奇霉素溶液的配制:称取阿奇霉素0.45g,溶于10ml的0.9%的生理盐水溶液中,配制成45mg/ml的溶液,待其充分溶解后,用0.22μm滤器过滤除菌后使用,每次使用时现用现配。溶液的配制及使用均应在无菌生物安全柜中操作。
b)硫酸阿托品溶液的配制:称取硫酸阿托品0.0425g,溶于10mL的0.9%的生理盐水溶液中,配制成4.25mg/ml的溶液,待其充分溶解后,用0.22μm滤器过滤除菌后使用,每次使用时现用现配。溶液的配制及使用均应在无菌生物安全柜中操作。
c)大环内酯类衍生物溶液的配制:称取适量大环内酯类衍生物,溶于10mL的0.9%的生理盐水溶液中,配制成0.05mol/L的溶液,待其充分溶解后,用0.22μm滤器过滤除菌后使用,每次使用时现用现配。溶液的配制及使用均应在无菌生物安全柜中操作。
d)琥珀酸普卡必利溶液的配制:称取琥珀酸普卡必利0.0009g,溶于10mL的0.9%的生理盐水溶液中,配制成0.09mg/ml的溶液,待其充分溶解后,用0.22μm滤器过滤除菌后使用,每次使用时现用现配。溶液的配制及使用均应在无菌生物安全柜中操作。
e)0.9%生理盐水的配置:称取0.9g NaCl,溶于100mL的无菌水中,用0.22μm的滤膜抽滤,溶液的配置及抽滤均应在无菌生物安全柜中操作。
实施例1大环内酯类衍生物的合成及鉴定
将红霉素A 28.58g(38.94mmol)、盐酸羟胺15.6g(224.4mmol)溶于60.0ml无水甲醇中,再加入三乙胺15.6ml(112.4mmol),搅拌加热回流24小时,反应完成后,0℃冷却抽滤,滤饼用少量甲醇洗涤,将所得固体悬浮于80ml甲醇中,搅拌下滴加20ml氨水,过滤除去不溶性物质,将甲醇氨水溶液滴加至150ml水中,有大量白色固体析出,抽滤,水洗至中性,干燥得到固体a(见图1),产率为80.6%,熔点为163-166℃,质谱数据为MS(M/Z):749.75(M+H+)。
将9-肟红霉素A(a)0.1g(0.134mmol)溶于1.6ml的丙酮中,在0-5℃温度下,边搅拌边滴加0.122g/ml的对甲苯磺酰氯溶液(溶剂为丙酮)0.416ml(0.269mmol)和0.045g/ml的碳酸氢钠水溶液1ml(0.536mmol),2h内滴加完毕,在室温下继续反应2h。反应完成后,将反应液抽滤,减压蒸馏滤液以蒸除丙酮,加入10%氢氧化钠,将pH值调至10,用DCM萃取3次,合并有机相,有机相用水洗2次,然后用无水硫酸钠干燥。蒸除DCM,真空干燥,得白色固体(b)(见图1),产率为91.8%,熔点为132-135℃,质谱数据为MS(M/Z):731.7374(M+H+)。
在0℃冰浴条件下,将0.1g b(0.1435mmol)、0.088g硼氢化钠(2.333mmol)溶于1.2ml无水甲醇中,搅拌反应4h,然后撤掉冰浴,继续在室温下反应20h,反应完成后,通入二氧化碳至无白色固体生成,抽滤,滤饼用少量甲醇洗涤,减压蒸除甲醇,得白色固体。加入DCM溶解白色固体,有机相用10%碳酸氢钠洗涤2次,分液,有机相加入100ml水,在搅拌条件下用1mol/L的盐酸调节pH值为2-3(优选为2.5),搅拌20分钟后用10%氢氧化钠调节PH值至6-7(优选为6.5),弃去有机层。水层加入20ml DCM,用10%氢氧化钠将pH值调至11,搅拌15分钟,分液得到DCM溶剂,真空干燥,得白色固体(III-1)(见图1),产率为70.6%,熔点为125-128℃,质谱数据为MS(M/Z):735.70(M+H+);核磁共振氢谱数据为:1H NMR(CDCl3,500MHz)δ5.15(d,1H),4.89(dd,1H),4.59(dd,1H),4.40(d,1H),4.11(dq,1H),3.61(d,1H),3.48(ddq,1H),3.34(s,3H),3.27(d,1H),3.22(dd,1H),3.02(d,1H),3.00(dd,1H),2.72(dq,1H),2.42(ddd,1H),2.33(d,1H),2.28(s,6H),2.25(dq,1H),1.94(ddq,1H),1.86(ddq,1H),1.71(m,2H),1.65(d,1H),1.65(dd,1H),1.57(dd,1H),1.45(ddq,1H),1.39(dd,1H),1.36(d,3H),1.27(s,3H),1.25(s,3H),1.24(m,1H),1.22(d,3H),1.18(d,3H),1.16(d,3H),1.05(s,3H),1.02(d,3H),0.91(d,3H),0.88(t,3H)。
将10g阿奇霉素(13.61mol)溶于70ml的盐酸中,在室温下搅拌反应5小时,在冰浴下加入10%的氢氧化钠,调节pH值至9,有大量白色固体析出,加入DCM萃取3次,合并有机相,减压蒸除DCM,真空干燥,得白色固体(III-2)(见图1),产率为70.6%,质谱数据为MS(M/Z):590.41(M+H+);核磁共振氢谱数据为:1H NMR(400MHz,DMSO-d6)δ6.20(br.s.,1H),5.07(d,J=6.59Hz,1H),4.95(d,J=11.00Hz,1H),4.55(d,J=7.40Hz,1H),4.31(s,1H),4.19(s,1H),4.04(d,J=8.09Hz,1H),3.38-3.45(m,3H),3.28-3.33(m,1H),2.99-3.11(m,1H),2.67(q,J=7.00Hz,1H),2.45-2.48(m,1H),2.40(dd,J=4.50,10.00,12.00Hz,1H),2.30(dd,J=3.00,12.00Hz,1H),2.21(s,9H),2.09(q,J=7.30Hz,1H),2.04(t,J=12.00Hz,1H),1.68-1.82(m,2H),1.59(d,J=12.37Hz,1H),1.36-1.45(m,2H),1.27-1.39(m,1H),1.05-1.14(m,10H),0.97(s,3H),0.94(d,J=6.59Hz,3H),0.87(d,J=7.17Hz,3H),0.83(d,J=6.82Hz,3H),0.75(t,J=7.22Hz,3H)。
将2.5g阿奇霉素溶于200ml盐酸中,油浴加热至40℃,搅拌反应10小时,然后使用10%氢氧化钠调节pH值为10,并且搅拌15分钟,使用二氯甲烷萃取3次,收集有机相,使用无水硫酸钠干燥,减压蒸除二氯甲烷,真空干燥,得白色固体为大环内酯类衍生物(III-3)(见图1),产率为81.8%;质谱数据为MS(M/Z):434.67(M+H+);核磁共振氢谱数据为:1H-NMR(400MHz,DMSO)δ6.15(s,1H),5.01(dd,J=10.9,2.1Hz,1H),4.60(d,J=5.7Hz,1H),4.31(s,1H),4.15(d,J=8.4Hz,1H),3.74(d,J=4.7Hz,1H),3.46(d,J=8.4Hz,1H),3.39(dd,J=9.8,6.0Hz,1H),3.28(d,J=4.7Hz,1H),2.68(q,J=6.4Hz,1H),2.55–2.43(m,1H),2.31(dd,J=12.4,2.7Hz,1H),2.24(s,3H),2.11–1.95(m,2H),1.85–1.69(m,2H),1.47–1.30(m,3H),1.11(d,J=6.7Hz,3H),1.03(d,J=15.7Hz,6H),0.96(d,J=6.7Hz,3H),0.84(d,J=7.0Hz,6H),0.78(s,3H)。
将1.5g III-1(2.0535mmol)溶于120ml的盐酸中,在50℃下搅拌反应10小时,在冰浴下加入10%的氢氧化钠,调节pH值至9,有大量白色固体析出,加入DCM萃取3次,合并有机相,减压蒸除DCM,真空干燥,得白色固体(III-4)(见图1),产率为60.1%,质谱数据为MS(M/Z):420.47(M+H+),核磁共振氢谱数据为1H-NMR(400MHz,DMSO)δ6.15(s,1H),5.01(dd,J=10.9,2.1Hz,1H),4.60(d,J=5.7Hz,1H),4.31(s,1H),4.15(d,J=8.4Hz,1H),3.74(d,J=4.7Hz,1H),3.46(d,J=8.4Hz,1H),3.39(dd,J=9.8,6.0Hz,1H),3.28(d,J=4.7Hz,1H),2.68(q,J=6.4Hz,1H),2.55–2.43(m,1H),2.31(dd,J=12.4,2.7Hz,1H),2.11–1.95(m,2H),1.85–1.69(m,2H),1.47–1.30(m,3H),1.11(d,J=6.7Hz,3H),1.03(d,J=15.7Hz,6H),0.96(d,J=6.7Hz,3H),0.84(d,J=7.0Hz,6H),0.78(s,3H)。
将35mg阿奇霉素溶于3ml二氯甲烷中,在0℃条件下将79mg酰氯类化合物滴加入反应液中,之后加入三乙胺,在0℃条件下反应2小时后,在室温条件下反应过夜,次日减压蒸除二氯甲烷,得化合物(III-5),产率为25%,质谱数据为MS(M/Z):1069.62(M+H+);核磁共振氢谱数据为:1H-NMR(400MHz,DMSO)δ7.75–7.67(m,4H),7.13(d,J=8.8Hz,2H),7.07(d,J=5.8Hz,2H),6.15(s,1H),5.01(dd,J=10.9,2.1Hz,1H),4.92(d,J=2.3Hz,1H),4.60(d,J=5.7Hz,1H),4.31(s,1H),4.15(d,J=8.4Hz,1H),4.12–3.99(m,4H),3.74(d,J=4.7Hz,1H),3.46(d,J=8.4Hz,1H),3.39(dd,J=9.8,6.0Hz,1H),3.28(d,J=4.7Hz,1H),2.68(q,J=6.4Hz,1H),2.55–2.43(m,1H),2.31(dd,J=12.4,2.7Hz,1H),2.11–1.95(m,2H),2.06–1.94(m,2H),1.85–1.69(m,2H),1.47–1.30(m,3H),1.11(d,J=6.7Hz,3H),1.03(d,J=15.7Hz,6H),0.96(d,J=6.7Hz,3H),0.84(d,J=7.0Hz,6H),0.78(s,3H)。
实施例2大环内酯类衍生物的抑菌活性测试
实验方法:
1)取5μL大肠杆菌甘油菌,接种到5mL的液体LB培养基中,置于37℃摇床中,220rpm/min,过夜培养;
2)将培养过夜的大肠杆菌倒入250ml的LB培养基中,37℃,220rpm培养3小时;
3)3小时之后,将锥形瓶中的菌液均分到小试管中,每试管中5ml,在各试管中加入5μl同等摩尔量的衍生物,培养24小时,每间隔2-4小时测试OD600值,并计算抑菌率,抑菌率=(1-(衍生物的OD600值)/(DMSO空白组的OD600值))x100%。
抑菌活性测试结果:
大环内酯类衍生物的抑菌活性如表1,表2所示,其中表1是化合物的浓度为5.344μM下的抑菌率,表2是化合物的浓度为10.688μM下的抑菌率;表1及表2的结果表明大环内酯类衍生物的抑菌活性明显低于大环内酯类抗生素阿奇霉素,通过结构改造,合成的大环内酯类衍生物的抑菌活性明显降低。
表1.大环内酯类衍生物在5.344μM浓度下对大肠杆菌的抑菌率
表2.大环内酯类衍生物在10.688μM浓度下对大肠杆菌的抑菌率
实施例3大环内酯类衍生物药效学检测
3.1肠蠕动乏力模型小鼠的给药治疗:
将45只小鼠随机分为九组:正常组、模型对照组、阳性药琥珀酸普卡必利组、阿奇霉素组,大环内酯类衍生物化合物III-1、化合物III-2、化合物III-3、化合物III-4、化合物III-5五个组,每组5只。正常组每只灌胃给予0.1mL的0.9%生理盐水;造模期间,模型对照组、大环内酯类衍生物组、阳性药琥珀酸普卡必利组和阿奇霉素组灌胃给予0.1mL的硫酸阿托品的水溶液,一天两次,连续给药两天。在建模后第1天,给予小鼠药物治疗,正常组以及对照组每次用0.1mL的0.9%的生理盐水灌胃,大环内酯类衍生物组每次分别用0.1mL的上述配置的0.05mol/L的大环内酯类衍生物水溶液灌胃,阳性药琥珀酸普卡必利组每次分别用0.1mL的上述配置的琥珀酸普卡必利溶液灌胃,阿奇霉素组每次分别用0.1mL的上述配置的阿奇霉素溶液灌胃,一天一次,给药一天。次日给碳末,1.5h后剖杀,观察并计算小鼠肠内的碳末推进距离,分别记录整段小肠的长度和碳末推进的小肠长度,并计算肠道碳末推进率,炭末推进率=(小肠全长-碳末推进的小肠长度)/小肠全长×100%。
3.2实验结果
通过观察实验中小鼠的生存状况,给药后小鼠均未出现中枢性毒副作用,如恶心呕吐等。
小鼠肠炭末推进率的结果如图2及表3所示:与肠蠕动乏力模型对照组小鼠相比,所制备的大环内酯类衍生物化合物III-1、化合物III-2、化合物III-3、化合物III-4、化合物III-5均具有不同程度的改善小鼠的肠蠕动乏力,促进小鼠的胃肠道动力的效果,其中化合物III-3改善效果好于改构前的化合物阿奇霉素,并且化合物III-3改善效果好于阳性药琥珀酸普卡比利。所制备的大环内酯类衍生物具有增强肠蠕动乏力模型小鼠的胃肠蠕动的能力。
表3.大环内酯类衍生物的炭末推进率
实施例4化合物III-3对家兔、比格犬、绒猴肠蠕动作用的检测
4.1实验方法
选择三种测试动物:家兔、比格犬、绒猴。根据体表面积换算法,依次计算出灌胃剂量为31.14mg/kg、17.8mg/kg、10mg/kg。收集灌胃前最后一次与灌胃后第一次粪便,将粪便进行称量和计数。
4.2实验结果
通过比较灌胃前与灌胃后动物粪便的重量和数量,发现给药化合物III-3后,可以显著增加实验动物的粪便重量和数量;同时给药组所排粪便便型较软,没有出现水样便。以上实验表明化合物III-3可以显著增强家兔、比格犬、绒猴的肠蠕动能力,具有促进肠道动力效果。
实施例5.化合物III-3大鼠的急毒实验
5.1实验方法
采用上下法估算化合物的LD50,具体方法如下:
(1)取大鼠1只,在1.75、5.5、17.35、55、175、550、2000mg/kg剂量范围内选择一个给药剂量,本试验选择175mg/kg灌胃给药。
(2)观察大鼠48h的存活状态,如果大鼠存活,第二只大鼠给予高一级剂量,即550mg/kg;如果大鼠死亡或出现濒死状态,第二只动物给予低一级的剂量,即55mg/kg,以此类推。本次试验大鼠在175、550、2000mg/kg剂量时全部存活且状态良好,考虑进行2000mg/kg剂量水平的限度试验。
(3)大鼠2000mg/kg剂量水平的限度试验。将化合物III-3给予1只大鼠。如果该动物死亡,则进行第(2)步实验;如果大鼠存活,依次将化合物给予另外4只大鼠,动物总数为5只。连续观察14天,如果1只大鼠在实验后期死亡,而其他大鼠存活,应停止对其他大鼠给药,对所有大鼠进行观察,是否在相似的观察期间也发生死亡。后期死亡的大鼠应与其他死亡大鼠同样计数,对结果进行评价如下:有3只或3只以上大鼠死亡时,LD50小于2000,;有3只或3只以上大鼠存活时,LD50大于2000;如果有3只以上大鼠死亡,则进行第(2)步试验。
5.2实验结果
给予SD大鼠2000mg/kg剂量的化合物后,在15天内,大鼠均未死亡,且未出现异常反应。表明其LD50大于2000mg/kg,化合物III-3急性毒性低,安全性好。
实施例6.化合物III-3号对大鼠心脏毒性的影响
6.1实验方法
将6只SD大鼠随机分为两组:正常组、给药大环内酯类衍生物化合物III-3组,每组3只。将大鼠麻醉后,正常组每只静脉注射0.5mL的0.9%生理盐水;大环内酯类衍生物组每只静脉注射0.5mL的1mM大环内酯类衍生物化合物III-3,心电信号采集使用ECG100C放大器,动物用的针式电极插入动物的皮下,在插入电极的时候,把要插入部位的皮肤提起来,插入电极后再放下皮肤。放大器增益设置在2000,高通滤波器设置在0.05Hz,低通滤波器设置在35Hz OFF。电极连接方法:VIN+连接左下肢,VIN-连接右上肢,GND连接右下肢。设置好心电信号的原始通道和采集参数后,开始采集心电信号.
6.2实验结果
通过观察实验中大鼠的心电图情况,给药化合物III-3后大鼠呼吸正常,行为未出现异常反应,给药组3只大鼠与对照组相比均未出现心率加快、心率不齐等心脏毒性现象。表明化合物III-3没有心脏毒性。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!