一种巯基功能化芳基羧酸的制备方法与流程
本发明涉及化合物合成技术领域,尤其涉及一种巯基功能化芳基羧酸的制备方法。
背景技术:
金属有机骨架材料(Metal-Organic framework,MOF)作为新兴的多孔材料得到了快速发展,该类多孔材料能够通过对功能配体的设计,进而定向自组装出具有预设结构和功能的金属有机框架材料。
基于前期的研究发现,巯基芳香羧酸化合物兼并了软硬原子为一体(软硬酸碱理论),其中羧基上的O作为硬碱,巯基上的S作为软碱,使这类化合物的优势十分显著。当选择硬酸类金属离子如(Zr4+)作为配位中心,中心离子会选择与羧基形成金属有机框架,而巯基不参与配位,游离并修饰骨架材料巯基功能化芳基羧酸化合物。巯基引入到金属有机骨架材料中,其优势主要体现如下:1)巯基官能团在有机方面的活泼性有利于MOF材料进行后合成修饰;2)巯基官能团和金属离子的强结合作用使得MOF材料能够吸收各种不同的金属离子从而达到去除重金属离子的目的。
除此以外,巯基化合物还是合成医药的中间体和重要的化工原料,主要有硫醇和硫酚两大类。巯基化合物的巯基易与汞离子、银离子等重金属快速络合,并形成稳定的配位化合物。因此,含巯基官能团的化合物在吸附、分离贵金属和重金属方面有巨大的应用前景。
目前,合成巯基化合物的反应,条件苛刻且剧烈,产率不高,操作繁琐,所用原料成本高,毒性大且具有十分强烈的刺激性气味等因素限制了巯基化合物的规模化生产。
如Jun He等(CN104447452A)报道了一种合成巯基功能化多芳基羧酸化合物的方法,反应式如式1:
该方法反应条件比较剧烈(反应温度较高),转化率及产率不高,反应时间较长,而且该方法所需原料(甲硫醇钠,酰氯化合物)价格比较昂贵,毒性大且具有十分强烈的刺激性气味,对操作人员的身体造成很大的危害,且不利于环境保护及工业化生产。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种巯基功能化芳基羧酸的制备方法,反应条件温和,且原料成本低。
本发明提供了一种巯基功能化芳基羧酸的制备方法,包括以下步骤:
A)对2,3,5,6-四氟对苯二甲酸的羧基进行保护,得到中间体1;
B)将中间体1和巯基酸酯在碱金属盐存在的条件下进行取代反应,得到中间体2;
C)中间体2进行水解反应,得到巯基功能化芳基羧酸。
优选的,所述步骤A)具体为:
2,3,5,6-四氟对苯二甲酸在甲醇与浓硫酸的条件下,进行酯化反应,得到2,3,5,6-四氟对苯二甲酸甲酯。
优选的,所述酯化反应的温度为回流,时间为1~24h。
优选的,所述巯基酸酯为3-巯基己醇己酸酯、巯基乙酸甲酯、3-巯基丙酸-3-甲氧丁酯、2-巯基丙酸丙酯、3-巯基丙酸-2-乙己酯中的一种或多种。
优选的,所述碱金属盐为磷酸钾、碳酸钠、乙酸钠、碳酸铯、碳酸钾、乙酸钾中的一种或多种。
优选的,所述中间体1、巯基酸酯和碱金属盐的摩尔比为1:(0.5~10):(1~20)。
优选的,所述取代反应的溶剂为四氢呋喃、异丙醇、二甲亚砜、乙腈、N,N-二甲基甲酰胺、1,3-二甲基-2-咪唑啉酮、聚乙二醇600、N,N-二甲基乙酰胺中的一种或多种。
优选的,所述取代反应的温度为0~80℃,反应时间为0.5~12h。
优选的,所述取代反应为一取代反应,二取代反应,三取代反应或四取代反应。
优选的,所述水解反应在强碱性条件下进行。
与现有技术相比,本发明提供了一种巯基功能化芳基羧酸的制备方法,包括以下步骤:A)对2,3,5,6-四氟对苯二甲酸的羧基进行保护,得到中间体1;B)将中间体1和巯基酸酯在碱金属盐存在的条件下进行取代反应,得到中间体2;C)中间体2进行水解反应,得到巯基功能化芳基羧酸。本发明采用巯基酸酯作为反应原料,成本较低,且毒性低,几乎没有气味,安全环保;并且反应条件温和,几乎没有能耗,反应迅速,节能环保,为工业化生产提供了可能;同时该反应具有较高的转化率和收率。
附图说明
图1是实施例1中间体2的核磁氢谱图;
图2是实施例1中间体2的核磁氟谱图;
图3是实施例1产物的核磁氢谱图;
图4是实施例1产物的核磁氟谱图;
图5是实施例1产物的质谱图。
具体实施方式
本发明提供了一种巯基功能化芳基羧酸的制备方法,包括以下步骤:
A)对2,3,5,6-四氟对苯二甲酸的羧基进行保护,得到中间体1;
B)将中间体1和巯基酸酯在碱金属盐存在的条件下进行取代反应,得到中间体2;
C)中间体2进行水解反应,得到巯基功能化芳基羧酸。
本发明首先对2,3,5,6-四氟对苯二甲酸的羧基进行保护,优选的,将2,3,5,6-四氟对苯二甲酸在甲醇与浓硫酸的条件下,进行酯化反应,得到2,3,5,6-四氟对苯二甲酸甲酯。
所述酯化反应的温度优选为回流,即50~100℃,反应时间优选为1~24h。
具体的,将原料2,3,5,6-四氟对苯二甲酸、甲醇与浓硫酸加入到带回流冷凝器的反应器中,开启搅拌,升温到回流1~24小时制备得到中间体1,即2,3,5,6-四氟对苯二甲酸甲酯。
反应结束后,对反应进行后处理,优选的,反应体系冷却至室温,加入大量蒸馏水,有大量固体析出,减压抽滤,得到白色片状晶体产物,即为中间体1。
然后将制备的中间体1和巯基酸酯在碱金属盐存在的条件下进行取代反应,得到中间体2。
所述巯基酸酯优选为3-巯基己醇己酸酯、巯基乙酸甲酯、3-巯基丙酸-3-甲氧丁酯、2-巯基丙酸丙酯、3-巯基丙酸-2-乙己酯中的一种或多种,更优选为一种或两种。
所述碱金属盐优选为磷酸钾、碳酸钠、乙酸钠、碳酸铯、碳酸钾、乙酸钾中的一种或多种。
所述中间体1、巯基酸酯和碱金属盐的摩尔比优选为1:(0.5~10):(1~20)。
所述取代反应的溶剂优选为四氢呋喃、异丙醇、二甲亚砜、乙腈、N,N-二甲基甲酰胺、1,3-二甲基-2-咪唑啉酮、聚乙二醇600、N,N-二甲基乙酰胺中的一种或多种。
所述取代反应的温度优选为0~80℃,反应时间优选为0.5~12h。
所述取代反应优选在惰性环境下进行。
具体的,将中间体1、巯基酸酯和碱金属盐加入反应器中,再将有机溶剂加入到反应器中,在惰性环境下0~80℃下反应0.5~12小时制备得到中间体2。
反应结束后,对反应进行后处理,优选的,反应体系经萃取得到有机层,然后进行柱色谱分离,得到无色粘稠液体,即为中间体2。
本发明中,所述取代反应可以通过控制巯基酸酯的用量,即通过改变中间体1与巯基酸酯的不同比例,控制实验进行取代氟原子(F)的一取代反应,二取代反应,三取代反应或四取代反应。从而可以使得此方法应用到含卤素原子(如F原子)的芳基化合物的巯基化反应。
当进行一取代反应时,所述中间体1与巯基酸酯的摩尔比为1:(0.1~5);
当进行二取代反应时,所述中间体1与巯基酸酯的摩尔比为1:(0.5~10);
当进行三取代反应时,所述中间体1与巯基酸酯的摩尔比为1:(1~15);
当进行四取代反应时,所述中间体1与巯基酸酯的摩尔比为1:(1~20)。
然后将中间体2进行水解反应,即可得到巯基功能化芳基羧酸。
优选的,所述水解反应在强碱性条件下进行。所述强碱性条件可以通过添加强碱性化合物得到,本发明对所述强碱性化合物并无特殊限定,可以为本领域技术人员熟知的强碱性化合物,本发明优选为KOH或NaOH。
所述水解反应优选在醇类溶剂和水的混合溶剂中进行。所述醇类溶剂优选为乙醇。
所述水解反应的温度优选为25~140℃,反应时间优选为0.5~24h。
反应结束后,对反应进行后处理,优选的,反应体系冷却至室温,加入过量浓盐酸,调节溶液至酸性,室温搅拌0.5~2h后,向体系中加入大量蒸馏水,有大量黄色固体析出,减压抽滤,得到黄色粉末状产物,即为巯基功能化芳基羧酸纯品,其收率高于90%。
上述反应的反应方程式如下:
其中,Base为碱金属盐;Solvent表示有机溶剂;x=1,2,3…….n,更优选的,x为1~10的整数。
本发明采用巯基酸酯作为反应原料,成本较低,且毒性低,几乎没有气味,安全环保;并且反应条件温和,几乎没有能耗,反应迅速,节能环保,为工业化生产提供了可能;同时该反应具有较高的转化率和收率,制备的巯基功能化芳基羧酸具有较高的纯度。
为了进一步说明本发明,下面结合实施例对本发明提供的巯基功能化芳基羧酸的制备方法进行详细描述。
实施例1
中间体1的合成步骤:
(1)称取原料2,3,5,6-四氟对苯二甲酸(1190mg,5mmol)加入100mL干燥的单口圆底烧瓶中。
(2)用量筒量取甲醇(无水,50mL)加入到单口圆底烧瓶中。
(3)滴加浓硫酸(2mL),室温下搅拌10min。然后置于油浴中回流24h。待反应完全后,冷却至室温,向混合物中加入大量的蒸馏水,有大量固体析出,减压抽滤,得到白色片状晶体产物(即中间体1)1237mg,收率93%,纯度98%。
中间体2的合成步骤:
(1)将碳酸钾(2208mg,16mmol)置于25mL干燥的反应茄瓶中,在氮气保护下使用注射器将3-巯基丙酸-2-乙基己酯(1.9mL,8.3mmol)打进反应茄瓶中。
(2)将在氮气下鼓泡10min后的N,N-二甲基甲酰胺(无水,15mL),通过真空管转移至25mL干燥的反应茄瓶中。
(3)最后,在氮气保护下,将上述制得的中间体1(1064mg,4mmol)加入到25mL干燥的反应茄瓶中,开启搅拌0.5~12h。
(4)反应完全后,萃取得到的有机层,经硅胶层析柱分离提纯,得无色粘稠液体产物(即中间体2)1903mg,收率72%,纯度98%。
对中间体2的结构进行检测,结果见图1和图2,图1是中间体2的核磁氢谱图,图2是中间体2的核磁氟谱图。
由图1可以看出,实施例1进行了二取代反应。
终产物3的合成步骤:
于24mL氢氧化钾的乙醇水溶液中加入中间体2(1800mg,2.72mmol),置于90℃的油浴中回流24h。反应完毕,冷却至室温,加入过量的浓盐酸,使溶液呈酸性。室温搅拌1h后,向混合溶液中加入大量的蒸馏水,有大量黄色固体析出,减压抽滤,得黄色粉末产物(即终产物3)658mg,收率91%,纯度97%。
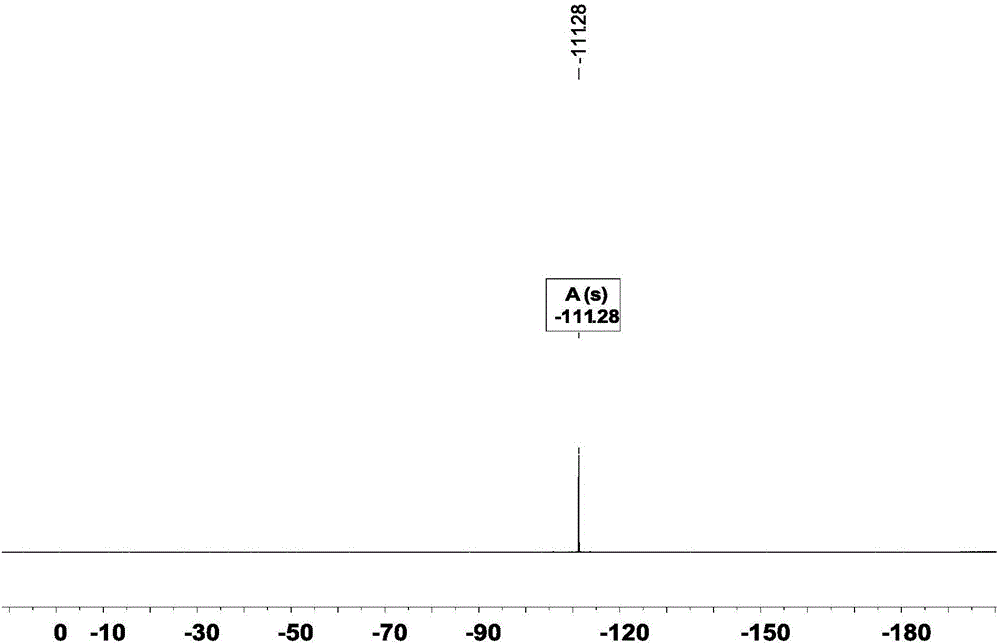
对产物结构进行检测,检测结果见图3和图4,其中,图3是产物的核磁氢谱图,图4是产物的核磁氟谱图。
由图3和图4可以看出,核磁氢谱图中,没有发现H的位移。原因是羟基和巯基上的氢比较活泼,会很容易被交换掉,在图谱上显现不出来。可见,中间体2的侧链取代基被水解掉。
采用质谱仪对分子结构进行检测,其质谱图见图5。
实施例2
中间体1的合成步骤:
(1)称取原料2,3,5,6-四氟对苯二甲酸(952mg,4mmol)加入80mL干燥的单口圆底烧瓶中。
(2)用量筒量取甲醇(无水,30mL)加入到单口圆底烧瓶中。
(3)滴加浓硫酸(1mL),室温下搅拌10min。然后置于油浴中回流24h。待反应完全后,冷却至室温,向混合物中加入大量的蒸馏水,有大量固体析出,减压抽滤,得到白色片状晶体产物(即中间体1)968mg,收率93%,纯度98%。
中间体2的合成步骤:
(1)将乙酸钾(1568mg,16mmol)置于25mL干燥的反应茄瓶中,在氮气保护下使用注射器将2-巯基丙酸丙酯(1228.4mg,8.3mmol)加入反应茄瓶中。
(2)将在氮气下鼓泡10min后的N,N-二甲基甲酰胺(无水,15mL),通过真空管转移至25mL干燥的反应茄瓶中。
(3)最后,在氮气保护下,将上述制得的中间体1(1064mg,4mmol)加入到25mL干燥的反应茄瓶中,开启搅拌0.5~12h。
(4)反应完全后,萃取得到的有机层,经硅胶层析柱分离提纯,得无色粘稠液体产物(即中间体2)1830mg,收率69%,纯度97%。
终产物3的合成步骤:
于20mL氢氧化钾的乙醇水溶液中加入中间体2(1800mg,2.72mmol),置于80℃的油浴中回流24h。反应完毕,冷却至室温,加入过量的浓盐酸,使溶液呈酸性。室温搅拌1h后,向混合溶液中加入大量的蒸馏水,有大量黄色固体析出,减压抽滤,得黄色粉末产物(即终产物3)600mg,收率83%,纯度97%。
通过核磁、质谱对产物结构进行检测,结果表明,本发明制备得到了2,5-二巯基-3,6-二氟对苯二甲酸。
实施例3
中间体1的合成步骤:
(1)称取原料2,3,5,6-四氟对苯二甲酸(1190mg,5mmol)加入100mL干燥的单口圆底烧瓶中。
(2)用量筒量取甲醇(无水,50mL)加入到单口圆底烧瓶中。
(3)滴加浓硫酸(2mL),室温下搅拌10min。然后置于油浴中回流24h。待反应完全后,冷却至室温,向混合物中加入大量的蒸馏水,有大量固体析出,减压抽滤,得到白色片状晶体产物(即中间体1)1237mg,收率93%,纯度98%。
中间体2的合成步骤:
(1)将碳酸铯(3258mg,10mmol)置于25mL干燥的反应茄瓶中,在氮气保护下使用注射器将3-巯基丙酸-2-乙基己酯(1.9mL,8.3mmol)打进反应茄瓶中。
(2)将在氮气下鼓泡10min后的N,N-二甲基乙酰胺(无水,15mL),通过真空管转移至25mL干燥的反应茄瓶中。
(3)最后,在氮气保护下,将上述制得的中间体1(1064mg,4mmol)加入到25mL干燥的反应茄瓶中,开启搅拌0.5~12h。
(4)反应完全后,萃取得到的有机层,经硅胶层析柱分离提纯,得无色粘稠液体产物(即中间体2)1882mg,收率71%,纯度97%。
终产物3的合成步骤:
于24mL氢氧化钠的乙醇水溶液中加入中间体2(1800mg,2.72mmol),置于90℃的油浴中回流24h。反应完毕,冷却至室温,加入过量的浓盐酸,使溶液呈酸性。室温搅拌1h后,向混合溶液中加入大量的蒸馏水,有大量黄色固体析出,减压抽滤,得黄色粉末产物(即终产物3)658mg,收率91%,纯度97%。
通过核磁、质谱对产物结构进行检测,结果表明,本发明制备得到了2,5-二巯基-3,6-二氟对苯二甲酸。
由上述实施例可知,本发明制备得到了巯基功能化芳基羧酸,获得了较高的收率和纯度。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!