稳定氢氰化催化剂的方法与流程
本申请是pct国际申请日为2013年5月24日,pct国际申请号为pct/us2013/0442640、中国国家申请号为201380028967.4的发明名称为《稳定氢氰化催化剂的方法》的申请的分案申请。
相关申请的交叉引用
本申请要求2012年6月1日提交的美国临时申请号61/654,584的优先权。本申请通过引用将该临时申请整体结合于此。
本公开涉及用于稳定镍-亚磷酸酯催化剂的方法,所述镍-亚磷酸酯催化剂在由烯烃制造腈中以及在使用用于进行亚磷酸酯向膦酸酯的转化的镍-亚磷酸酯催化剂的方法中使用。
背景技术:
己二腈(adn)在可用于形成薄膜、纤维和模制品的尼龙聚酰胺的工业生产中是一种商业上重要并且多用途的中间体。adn可以通过1,3-丁二烯(bd)在包含各种含磷配体的过渡金属配合物的存在下的氢氰化而生产。例如,在现有技术中很好地记载了包含镍和单齿含磷配体的催化剂;参见,例如,美国专利号3,496,215;3,631,191;3,655,723和3,766,237;以及tolman,c.a.,mckinney,r.j.,seidel,w.c.,druliner,j.d.,和stevens,w.r.,advancesincatalysis(催化进展),1985,第33卷,1-46页。也公开了烯键式不饱和化合物用包含镍和某些多齿亚磷酸酯配体的催化剂的氢氰化的改进;例如,参见:美国专利号5,512,696;5,821,378;5,959,135;5,981,772;6,020,516;6,127,567;和6,812,352。
活化烯烃如共轭烯烃(例如,1,3-丁二烯)的氢氰化可以在不使用路易斯酸助催化剂的情况下以可用的速率进行。然而,对于直链腈如adn的生产来说,未活化烯烃如3-戊烯腈(3pn)的氢氰化需要至少一种路易斯酸助催化剂以获得工业上可用的速率和产率。例如,美国专利号3,496,217、4,874,884、和5,688,986公开了路易斯酸助催化剂用于非共轭烯键式不饱和化合物用包含含磷配体的镍催化剂的氢氰化的应用。作为结果,在bd向adn的两步转化中,可以在不存在路易斯酸助催化剂的情况下进行将bd转化为3pn的第一氢氰化步骤,而通过使用路易斯酸,例如zncl2,促进将3pn转化为adn的第二氢氰化步骤。典型地,例如,为了避免亚磷酸酯配体水解,此类反应与在实践中一样在完全不含水的情况下进行。
如由本文的申请人在共同未决申请中公开的,用于3-戊烯腈的氢氰化的方法可以包括:在水,优选受控浓度的水的存在下,将3-戊烯腈和hcn进料至包含路易斯酸助催化剂、镍和含磷配体的氢氰化反应区。在通过引用整体结合在本文中的那个申请中,公开了意外的发现:通过在通过催化剂回收过程再循环催化剂时维持反应混合物中水的特定浓度,经过比具有更高或更低,即接近零,的浓度的水的其他过程多的再循环循环次数,氢氰化催化剂存量维持其活性。在其中公开了:由于在其中描述的各种原因,发现有利的是维持反应区内的水浓度范围,这足以在连同下游液-液萃取和催化剂配合物的再循环的连续操作下提高催化剂存量的活性。
然而,尽管提高了催化剂存量的活性,氢氰化反应区中水的存在可能会导致镍催化剂的某些亚磷酸三芳基酯配体的水解以产生酸性亚磷酸酯配体水解产物(lhp),如由亚磷酸三芳基酯的水解产生的亚磷酸二芳基酯,其可以经历进一步降解成为亚磷酸单芳基酯和亚磷酸。因为配体水解过程本身可以是酸催化的,酸性lhp的累积可能会进一步提高配体水解的速率,从而降解活性镍-亚磷酸酯氢氰化催化剂。
技术实现要素:
本文的发明人已经意外地发现,通过将如在本文中所公开的二齿亚磷酸酯配体加入至包含镍-亚磷酸酯催化剂的氢氰化环境,或者通过使用包含如在本文中所公开的二齿配体的镍-亚磷酸酯催化剂,可以降低或消除酸性配体水解产物(lhp)的累积以及所造成的镍-亚磷酸酯催化剂的加速分解,所述镍-亚磷酸酯催化剂用于氢氰化反应,例如,丁二烯的氢氰化或戊烯腈的氢氰化,如在路易斯酸、氰化氢、和水的存在下。本申请涉及将酸性lhp亚磷酸二芳基酯转化为不催化镍-亚磷酸酯催化剂的亚磷酸酯配体的降解的中性膦酸二酯产物的方法。本发明还涉及此类转化的中性膦酸酯产物。本发明还涉及戊烯腈氢氰化反应,其中抑制了配体降解并且延长了催化剂寿命;并且本发明还涉及稳定用于在存在水的条件下的戊烯腈氢氰化的镍-亚磷酸酯催化剂的方法。
附图说明
图1显示说明据信作为配体水解产物(lhp)的形成和失活的反应的方案;上面的反应说明了lhp的形成,并且下面的反应说明了酸性lhp的烷基化失活。
图2显示说明据信作为由亚磷酸二芳基酯lhp形成氰基丁基膦酸酯的机制的反应方案。
图3显示说明据信作为由亚磷酸二芳基酯lhp形成丁烯基膦酸酯的机制的反应方案。
图4是显示在戊烯腈中在水并且在48小时zncl2的存在下由配体2水解得到的,lhp形成随时间的时间进程的图表。
图5显示在75℃、在500ppmh2o的存在下、在二齿配体3的存在下静置5天后的催化剂配体副产物的gc/ms痕量。
图6是显示在配体3-镍配合物的存在下配体1、配体2、和配体3的混合物的水解速率的图表。参见实施例1。
图7是显示实施例1的对照实验的结果的图表,其中在不存在镍的情况下,配体1、配体2和配体3的水解以及lhp和副产物2,4x的形成表现出诱导期,之后加速的亚磷酸酯水解速率,其为自催化反应的特征。
图8显示了在配体4-镍配合物的存在下配体2和配体4的混合物的水解的时间进程的图表。
图9是显示在戊腈中配体3和配体1的混合物的水解的时间进程的图表,其中在混合物中存在由配体3形成的镍亚磷酸酯配合物。
具体实施方式
说明书中提及的“实例”、“实施方案”等表示所描述的实施方案的实例可以包括特定的特征、结构、或特性,但是每个实例或实施方案可以不必须包括该特定的特征、结构、或特性。此外,此类短语并非必须指代相同的实例或实施方案。此外,当与实例或实施方案一起描述特定的特征、结构、或特性时,应该认为无论是否明确地说明,与其他实例或实施方案一起实施这种特征、结构、或特性都是在本领域技术人员的知识范围之内的。
以范围格式表述的值应该以灵活的方式解释为不仅包括作为范围的界限而明确叙述的数值,而且也包括在那个范围内的所包含的所有单个数值或子范围,如同每个数值和子范围被明确地叙述一样。例如,“约0.1%至约5%”的浓度范围应当被解释为:不仅包括明确叙述的浓度约0.1重量%至约5重量%,而且也包括所指出的范围内的单个浓度(例如,1%、2%、3%和4%)和子范围(例如,0.5%、1.1%、2.2%、3.3%和4.4%)。
在本文献中,除非另外指出,术语“一个”或“一种”用于包括一或多于一,并且术语“或”用于指代包含的“或”。此外,应该理解的是,在本文中采用的且没有另外定义的措词或术语仅用于描述目的而不是限制目的。
如在本文中所使用的短语如“在适合提供……的条件下”或“在足以产生……的条件下”等在合成方法的上下文中是指反应条件,如时间、温度、溶剂、反应物浓度等,其在实验者的常规技术内改变,提供反应产物的可用数量或产率。所需的反应产物不必是唯一的反应产物,或者起始材料不必完全消耗,只要能够分离或另外使用所需的反应产物即可。
通过“化学上可行的”,意指在其中不违背一般理解的有机结构规则的结合排列方式或化合物;例如在某些情况下将包含不会自然存在的五价碳原子的权利要求的定义内的结构应理解为不在权利要求内。在本文中所公开的结构以其全部特征意在仅包括“化学上可行的”结构,并且在本文中不意图公开或要求保护任何叙述的非化学上可行的结构,例如在示出具有可变原子或基团的结构中。
结构的所有手性的、非对映的、外消旋的形式是预期的,除非具体指定特定的立体化学或异构形式。在多个实例中,尽管在特别要求保护的化合物中描述了个别立体异构体,立体化学名称并非意味着备选的异构形式是次优选的、不希望的、或不要求保护的。如从描述中显而易见的,本发明中使用的化合物可以在任一或所有非对称原子处以任何富集程度包含富集的或拆分的光学异构体。可以分离或合成外消旋和非对映混合物二者以及单独的光学异构体,从而基本不含其对映体或非对映体,并且这些全部在本发明的范围内。
如在本文中所使用的,术语“稳定化合物”和“稳定结构”意指表示足够强以至于可以经受从反应混合物中分离为可用纯度并且形成为有效治疗剂的化合物。本文仅考虑稳定化合物。
当在没有对基团中原子数任何限制的情况下提及基团例如“烷基”时,应理解的是,在烷基的大小方面限定并限制权利要求,其借助以下两者:借助定义;即,基团如烷基所具有的大小(碳原子数)是有限的数量,其小于宇宙中的碳原子总数并且受本领域普通技术人员理解的约束以使基团的大小对于分子实体来说是合理的;并且借助官能度,即基团如烷基的大小受该基团安置于含有该基团的分子上的官能性如在水性或有机液体介质中的溶解度的约束。因此,叙述“烷基”或其他化学基团或部分的权利要求是受限的并且受约束的,因为基团中的原子数不能是无限的。
此外,在以马库什组描述本发明的特征或方面的情况下,本领域技术人员将会理解,由此也以马库什组的任何单个成员或成员的子组描述本发明。例如,如果将x描述为选自由溴、氯、和碘组成的组,则充分描述了x是溴的权利要求和x是溴和氯的权利要求。此外,在以马库什组描述本发明的特征或方面的情况下,本领域技术人员将会理解,由此也以马库什组的单个成员的任何组合或成员的子组描述本发明。因此,例如,如果将x描述为选自由溴、氯、和碘组成的组,并且将y描述为选自由甲基、乙基、和丙基组成的组,则充分描述了x是溴并且y是甲基的权利要求。
如果将必须为整数的变量,例如烷基中的碳原子数或环上取代基的数量的值描述为范围,例如0-4,则意指该值可以是0和4(含)之间的任何整数,即0、1、2、3、或4。
化合物或化合物的组,如在本发明的方法中使用的,可以包括在本文中列出的特征的任何组合和/或子组合中的任一个。
也提供了如在任一实施例中或示例性化合物中所示的化合物。
限制性条件可以适用于公开的类目或组,其中可以从这些类目或组中排除其他公开的类目、组、特征、化合物或物种中的任何一个或多个。
作为在本文中使用的术语的“芳基”是指在环中不含杂原子的环状芳族烃。因此芳基包括,但不限于,苯基、萘基、薁基、庚搭烯基、联苯基、联萘基、引达省基(indacenyl)、芴基、菲基、三亚苯基、芘基、并四苯基、
作为在本文中使用的术语的烷基是指具有1至约20个碳原子并且典型1至12碳或1至8个碳原子的直链和支链烷基。直链烷基的实例包括具有1至8个碳原子的那些直链烷基如甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、和正辛基。支链烷基的实例包括,但不限于,异丙基、异丁基、仲丁基、叔丁基、新戊基、异戊基、和2,2-二甲基丙基。如在本文中所使用的,术语“烷基”包括正烷基、异烷基、和反异烷基(anteisoalkyl)基团以及其他支链形式的烷基。
作为在本文中使用的关于亚磷酸酯配体的术语,术语“单齿”和“二齿”分别是指包含一个或两个与金属配位的磷原子的分子实体。单齿和二齿的亚磷酸酯二者与镍原子形成配合物,所述配合物如在路易斯酸和水的存在下可以催化氢氰化反应,例如戊烯腈成为己二腈的氢氰化反应。
作为在本文中使用的术语的单齿亚磷酸酯配体的实例包括亚磷酸三芳基酯结构,如下式的化合物
其中每个ar表示芳基,其可以是相同的或不同的。例如,全部三个ar基团可以是2,4-二甲苯基,从而提供下式的单齿配体,
其适合金属如镍以各种氧化态的配位。在这种类型的分子实体中,每个磷原子各自经由相应的氧原子结合到三个取代的苯基环,即通过三个单官能芳基。
作为在本文中使用的术语的单齿亚磷酸酯配体的额外的实例包括含有单个磷原子的环状亚磷酸酯,如下式的化合物
在这种类型的分子实体中,每个磷原子经由氧原子结合到取代的苯基,并且经由两个相应的氧原子结合到取代的联苯基两次,即亚磷酸酯通过一个单官能芳基和一个双官能芳基酯化。
作为在本文中使用的术语的二齿亚磷酸酯配体是指每分子包含两个磷原子的分子实体。两个磷原子结合到双官能芳基,并且可以结合到额外的单官能或双官能芳基。实例是
另一个实例是
其中两个磷原子中的每一个结合到双官能芳基例如联苯基或联萘基,每个磷原子经由相应的氧原子结合,并且两个磷原子中的每一个进一步与两个相应的单官能芳基结合。
作为在本文中使用的术语的亚磷酸酯(盐)或亚磷酸酯是指包含一个或多个三价磷原子的化合物,每个磷原子与三个氧原子结合,氧原子中的每一个可以带有质子或者可以与碳部分结合。被称为“亚磷酸三芳酯”的亚磷酸三芳基酯是在其中三个氧原子中的每一个带有芳基并且具有式(aro)3p的化合物,其中各自独立选择的ar表示芳基。亚磷酸三芳基酯的一个实例是配体5,即以下通式的异构体的混合物
“亚磷酸二芳基酯”是式(aro)2poh的化合物,同样地,其中各自独立选择的ar表示芳基。如可以看到的,亚磷酸二芳基酯含有oh基团或其等同物,其包含酸性质子,这为亚磷酸二芳基酯赋予酸的性质。该术语表现为两种互变异构体形式,通常如下所示描绘:
无论氢原子的结合位置,此类化合物均具有酸性。
“亚磷酸单芳基酯”是式(aro)p(oh)2的化合物。游离oh基团或其等同物的存在也为亚磷酸单芳基酯赋予酸性。如在本领域内公知的,游离的酸是亚磷酸,h3po3。
作为在本文中使用的术语的“膦酸酯”,即膦酸的酯,包含磷-碳键、五价磷原子、和三个与磷原子结合的氧原子,其中两个可用于与质子或与碳部分形成另外的键。
如果两个可用的氧原子与碳部分结合,所得到的化合物是膦酸二酯,例如二芳膦酸基二酯r-p(=o)(oar)2;如果仅一个,则是膦酸单酯,例如膦酸单芳基酯r-p(=o)(oar)(oh)。游离的酸是膦酸,r-p(=o)(oh)2。尽管膦酸单酯和游离的酸具有酸性,膦酸二酯不具有可电离的质子,因此是非酸性的,被称为“中性”膦酸酯或膦酸二酯。
通过本发明的方法由酸性亚磷酸二芳基酯配体水解产物形成中性膦酸二酯产物涉及包括:利用在氢氰化反应混合物中存在的试剂如戊烯腈、丁二烯、丁二烯二聚物以及他们的氢氰化产物等直接烷基化亚磷酸二芳基酯lhp的磷原子以产生膦酸酯等。类似地,如果在氢氰化过程的反应环境中发生亚磷酸二芳基酯的进一步配体水解,本发明的发明人了解,也会将此类亚磷酸单芳基酯类似地转化为在其中酸性oh基团已经通过烷基化过程中和的中性物种。因此,通过使用本发明的方法,减少了能够催化催化剂配体水解的酸性lhp产物的形成。
如在本领域内公知的,“路易斯酸”是可以起电子受体的作用的化合物。各种金属卤化物如zncl2,是路易斯酸的实例。
本发明的一个方面是一种形成式(i)的膦酸二酯化合物的方法
其中
每个ar独立地为未取代的或单取代的或多取代的芳基,其中任意芳基可以被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基取代;或者,
所述两个ar基团彼此结合以提供未取代的或取代的联芳衍生物,其中所述联芳衍生物是未取代的或者独立地被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基单取代或多取代;并且,
r是2-丁烯基、3-丁烯基、2-氰基丁基、3-氰基丁基、或4-氰基丁基;
所述方法包括:在反应区中使式(iii)的亚磷酸二芳基酯化合物、戊烯腈、氰化氢、路易斯酸、和水在包含式(iv)的二齿亚磷酸酯配体的镍催化剂的存在下,在适合发生所述戊烯腈的氢氰化的条件下接触,
其中ar如对于式(i)的所述化合物所定义,
其中r1和r2各自独立地为未取代的或取代的单价芳基,并且r3-r10中的每一个独立地选自由氢、(c1-c10)烷基、和(c1-c10)烷氧基组成的组,或者其中两个相邻的r3-r10基团一起形成任选取代的稠合芳基环。
对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基,其中所述r1和r2芳基各自被一个相应的(c1-c10)烷基或(c1-c10)烷氧基邻位取代,条件是所述r1和r2芳基的相应间位和对位可以是未取代的或取代的。
对于式(iv)的二齿亚磷酸酯配体来说,r6和r10优选不是氢。
对于式(iv)的二齿亚磷酸酯配体来说,r3-r5中的至少一个以及r7-r9中的至少一个优选不是氢。
对于式(iv)的二齿亚磷酸酯配体来说,r5和r6一起可以任选地形成成(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环,并且其中r9和r10一起形成任选(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环。
对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基并且所述芳基是被一个相应(c1-c10)烷基或(c1-c10)烷氧基邻位取代的,条件是所述r1和r2芳基的相应间位和对位可以是未取代的或取代的;r6和r10是(c1-c10)烷基或(c1-c10)烷氧基,并且r3-r5中的至少一个以及r7-r9中的至少一个是(c1-c10)烷基或(c1-c10)烷氧基。
更具体地,戊烯腈可以是3-戊烯腈。
式(iv)的r1和r2还可以是2,4-二甲苯基。
r3和r7还可以是异丙基,r4和r8可以是氢,或r5、r6、r9和r10可以是甲基,或者它们的任何组合。
更具体地,式(iv)的二齿亚磷酸酯配体可以是
在与镍的催化配合物中,在路易斯酸zncl2的存在下,其中这些二齿磷酸酯配体存在于氢氰化反应区中的活性催化形式据信分别具有以下结构:
本发明还涉及一种式(i)的膦酸二酯化合物,
其中
每个ar独立地为未取代的或单取代的或多取代的芳基,其中任意芳基可以被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基取代;或者,
所述两个ar基团彼此结合以提供未取代的或取代的联芳基,其中所述联芳基是未取代的或者独立地被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基单取代或多取代;并且,
r是2-丁烯基、3-丁烯基、2-氰基丁基、3-氰基丁基、4-氰基丁基,或者是氰基壬烯基的异构体的组中的一种,其中所述异构体的组包括双键和氰基位置异构体。
r可以是2-丁烯基或3-丁烯基。r还可以是2-氰基丁基、3-氰基丁基、或4-氰基丁基,或者r可以是氰基辛烯基的异构体的组中的一种,其中所述异构体的组包括双键和氰基位置异构体。作为在本文中使用的术语的氰基辛烯基是指包含八个碳原子、含碳原子的氰基、和双键以任何可能的异构构型的基团,以提供具有总计九个碳原子、腈官能团、和不饱和的基团。例如,氰基辛烯基可以包括下式的基团
其中波浪线表示氰基辛烯基通过其结合到膦酸酯的磷原子的键,即8-氰基-辛-3-烯基。
对于式(i)的膦酸二酯化合物来说,两个ar基团可以是(c1-c4)烷基取代的苯基,或者两个ar基团合起来可以是(c1-c4)烷基多取代的联苯基,或者一个ar基团可以是(c1-c4)烷基取代的苯基并且一个ar基团可以是进一步被羟基取代的、(c1-c4)烷基多取代的联苯基。
式(i)的化合物可以选自由下列各项组成的组:
其中r是以上定义的,即2-丁烯基、3-丁烯基、2-氰基丁基、3-氰基丁基、4-氰基丁基、或氰基辛烯基异构体。
本发明还涉及一种用于生产己二腈的连续氢氰化方法,所述方法包括:使用包含镍和配体混合物的镍-亚磷酸酯催化剂,所述配体混合物包含一种或多种单齿亚磷酸酯配体和式(iva)的二齿亚磷酸酯配体
其中r1和r2二者均为单邻位取代的芳基并且所述芳基是被一个相应(c1-c10)烷基或(c1-c10)烷氧基邻位取代的,条件是所述r1和r2芳基的相应间位和对位可以是未取代的或取代的;r6和r10是(c1-c10)烷基或(c1-c10)烷氧基,r3-r5和r7-r9各自独立地为氢、(c1-c10)烷基或(c1-c10)烷氧基,条件是r3-r5中的至少一个以及r7-r9中的至少一个是(c1-c10)烷基或(c1-c10)烷氧基;其中所述方法表现为,所述催化剂的降解相对于在可比较的条件下第二镍-亚磷酸酯催化剂的降解的量减少,在所述可比较的条件中所述配体混合物仅包含一种或多种单齿亚磷酸酯配体,或者包含不包括式(iva)的化合物在内的二齿配体,所述方法包括:
(a)将所述催化剂、3-戊烯腈、路易斯酸、hcn、和水装入在适合发生氢氰化的条件下的反应区;
(b)从所述反应区中取出包含氢氰化产物和回收的催化剂的反应器流出物;
(c)使所述反应器流出物与萃取溶剂接触以从氢氰化产物中分离回收的催化剂;和
(d)将所述回收的催化剂的一部分和补充催化剂的一部分重新装入所述反应区,这足以在一定时间段内将活性催化剂的量与在所述时间段内加入的3-戊烯腈的量的比率维持基本恒定,
其中补充催化剂的相对部分与在进行可比较的过程中使用的补充催化剂的相对部分减少,在所述可比较的过程中所述配体混合物仅包含单齿亚磷酸酯配体或者包含不包括式(iva)的所述化合物在内的二齿配体。
在上述方法中,重新装入包含式(iva)的所述二齿亚磷酸酯配体的所述催化剂的所述反应区的补充催化剂的一部分小于装料至仅包含一种或多种单齿亚磷酸酯配体或者包含不包括式(iva)的所述化合物在内的二齿配体的所述催化剂的所述反应区的补充催化剂的一部分。
更具体地,式(iva)的二齿亚磷酸酯配体可以是配体3
或者可以是配体4
或它们的混合物,并且其中与在进行可比较的过程中发生的所述催化剂的降解相比,发生所述催化剂的减少的降解,在所述可比较的过程中所述配体混合物不包含配体3或配体4。
公开的本发明的氢氰化方法可以包括:控制反应区中水的量。所述方法适合连续运行以使催化剂配合物再循环。因此,例如,所述方法可以包括:使3-戊烯腈中在反应区中在包含含磷配体和镍金属的催化剂配合物的存在下氢氰化,其包括使3-戊烯腈、路易斯酸、hcn和受控量的水流动至反应区;从反应区中取出包含氢氰化产物和催化剂配合物的反应器流出物;使反应器流出物与萃取溶剂接触以回收催化剂配合物并且从催化剂配合物中移除杂质;以及使至少一部分回收的催化剂配合物再循环至反应区。所述方法可以包括:控制反应区中的水浓度以维持循环的催化剂存量中所需的催化剂组成。本文的发明人已经意外地发现,与其中不采用如在本文中所公开的式(iv)或的式(iva)的二齿配体的可比较的过程相比,使用如在本文中所公开的式(iv)或的式(iva)的二齿亚磷酸酯配体降低了在进行氢氰化过程期间的催化剂分解的速率。这使得补充催化剂的量降低,所述补充催化剂将会是维持反应区中活性催化剂的量与3-戊烯腈的量的基本恒定比率所需要的。
适合的水浓度的实例包括0.5ppm多至反应混合物的饱和极限,例如0.5ppm至约和约2000ppm,或约1ppm和1000ppm,或约5ppm至约500ppm,或约10ppm至约350ppm,或约20ppm至约300ppm,或为约240ppm。
与本领域普通技术人员理解的相反,在氢氰化反应中使用受控的水浓度是令人意外的,因为一般理解的是水引起催化剂配合物的降解,一般认为在反应混合物中应该将水的浓度维持得尽可能低,以通过重复的再循环循环使催化剂配合物的活性最大化。本文的发明人在共同未决专利申请中公开了该主题。
本文的发明人已经意外地发现,如可以进行以获得上述共同未决专利申请中描述的益处的,其中存在水的氢氰化过程中使用如在本文中所公开的式(iv)的或式(iva)的二齿配体可以抑制本身可能会催化进一步配体水解的酸性配体水解产物(lhp)在反应环境中的累积或浓度增加。这通过在镍-亚磷酸酯催化剂的存在下酸性配体水解产物向中性膦酸二酯化合物的催化转化而完成,其中存在本文中所公开的二齿配体,例如式(iv)的或式(iva)的二齿配体。本发明可以提供比其他方法更高效的在己二腈生产中使氢氰化3-戊烯腈的方法,例如,通过更高效地利用有价值的配体或衍生自配体的催化剂,包括通过经由含配体催化剂配合物的连续再循环将配体或由配体形成的催化剂的催化活性维持在较高的水平。
本发明还涉及一种用于如上所述的生产己二腈的连续氢氰化方法,其中式(iv)或的式(iva)的二齿亚磷酸酯配体是配体3或配体4,或它们的混合物,并且其中与在进行可比较的过程中存在的所述催化剂的降解相比,发生所述催化剂的减少的降解,在所述可比较的过程中所述配体混合物不包含配体3或配体4。
本发明还涉及一种将式(iii)的酸性亚磷酸酯配体水解产物转化为电中性膦酸酯形式的方法;
其中每个ar独立地为未取代的或单取代的或多取代的芳基,其中任意芳基可以被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基取代;或者,
所述两个ar基团彼此结合以提供未取代的或取代的联芳基,其中所述联芳基是未取代的或者独立地被独立选择的(c1-c4)烷基、羟基、亚磷酸酯基、或膦酸酯基单取代或多取代;
所述方法包括:在式(iv)的二齿亚磷酸酯配体的存在下
其中r1和r2各自独立地为未取代的或取代的单价芳基,并且r3-r10中的每一个独立地选自由氢、(c1-c10)烷基、和(c1-c10)烷氧基组成的组,或者其中两个相邻的r3-r10基团一起形成任选取代的稠合芳基环;
使式(iii)的所述配体水解产物和未取代或取代的烯烃在所述烯烃、氰化氢、和水的存在下,在氢氰化反应区中,在适合产生其电中性膦酸酯形式的条件下接触。
对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基,其中所述芳基r1和r2分别是被(c1-c10)烷基或(c1-c10)烷氧基单邻位取代的。芳基可以是间位和/或对位取代的,但是在这些情况下,根据定义,r1和r2中的每一个可以仅带有单个烷基或烷氧基取代基。
对于式(iv)的二齿亚磷酸酯配体来说,r6和r10优选不是氢。
对于式(iv)的二齿亚磷酸酯配体来说,r3-r5中的至少一个以及r7-r9中的至少一个优选不是氢。
对于式(iv)的二齿亚磷酸酯配体来说,r5和r6一起可以任选地形成成(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环,并且其中r9和r10一起形成任选(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环。
对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基并且所述芳基是被(c1-c10)烷基或(c1-c10)烷氧基邻位取代的,r6和r10是(c1-c10)烷基或(c1-c10)烷氧基,并且r3-r5中的至少一个以及r7-r9中的至少一个是(c1-c10)烷基或(c1-c10)烷氧基。
例如,r1和r2可以是2,4-二甲苯基。
例如,式(iv)的化合物可以是
更具体地,在所述方法中,烯烃可以是戊烯腈,如3-戊烯腈。
本发明还涉及一种针对在有效用于在路易斯酸和水的存在下戊烯腈的氢氰化的反应条件下水解配体分解而稳定氢氰化催化剂的方法,所述催化剂包含含有镍和一种或多种亚磷酸三芳基酯配体的镍-亚磷酸酯配合物,所述方法包括:将二齿亚磷酸酯配体加入至所述镍-亚磷酸酯配合物,所述二齿配体具有式(iv)
其中r1和r2各自独立地为未取代的或取代的单价芳基;r3-r10中的每一个独立地选自由氢和c1-10烷基组成的组,或者其中两个相邻的r3-r10基团一起形成任选取代的稠合芳基环。
例如,对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基,其中所述芳基是被(c1-c10)烷基或(c1-c10)烷氧基单邻位取代的。例如,r1和r2可以是2,4-二甲苯基。或者,对于式(iv)的二齿亚磷酸酯配体来说,r6和r10可以不是氢。
对于式(iv)的二齿亚磷酸酯配体来说,r3-r5中的至少一个以及r7-r9中的至少一个优选不是氢。或者,对于式(iv)的二齿亚磷酸酯配体来说,r5和r6一起可以任选地形成成(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环,并且其中r9和r10一起可以任选地形成(c1-c10)烷基或(c1-c10)烷氧基取代的稠合苯基环。
更具体地,对于式(iv)的二齿亚磷酸酯配体来说,r1和r2二者均可以为单邻位取代的芳基并且所述芳基可以是被(c1-c10)烷基或(c1-c10)烷氧基邻位取代的,r6和r10可以是(c1-c10)烷基或(c1-c10)烷氧基,并且r3-r5中的至少一个以及r7-r9中的至少一个可以是(c1-c10)烷基或(c1-c10)烷氧基。
更具体地,在本发明的方法中,戊烯腈是3-戊烯腈。
更具体地,式(iv)的二齿亚磷酸酯配体可以是
图1说明了在进行本发明的方法中发生的反应。包含具有配体如三-(2,4-二甲苯基)亚磷酸酯的镍配合物的催化剂,所示化合物经过与水在酸(h+)和戊烯腈(“pn”)的存在下的反应,产生如示出的游离的2,4-二甲苯酚和亚磷酸二芳基酯lhp。
在图1的第二步中,亚磷酸二芳基酯说明性地经过与2-戊烯腈在包含二齿配体(配体3)-镍配合物的催化剂的存在下反应,产生本发明的膦酸二酯,其是非酸性的并且不催化在水的存在下进一步的配体水解。2-戊烯腈、3-戊烯腈、和4-戊烯腈异构化合物据信在氢氰化反应的条件下是可互换的。
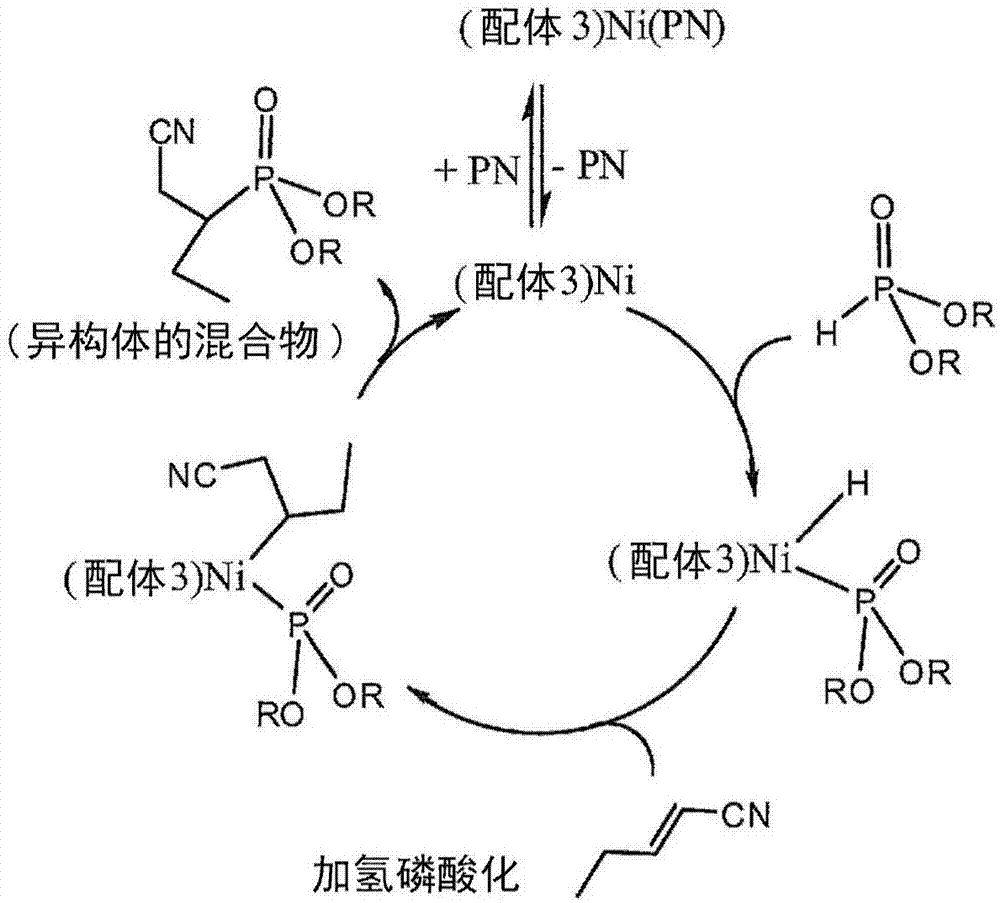
尽管不希望受理论的约束,图2说明了本文的发明人所相信的是由其实现了lhp失活和氰基丁基膦酸二酯形成的反应循环。亚磷酸二芳基酯lhp(即,其中r=芳基)据信与包含二齿配体(在这里,例如,配体3)的镍-亚磷酸酯催化剂配位,之后与戊烯腈(例如,2-戊烯腈)反应以产生氰基丁基膦酸酯的异构混合物。
图3说明了:除了与可以在戊烯腈的氢氰化的反应环境中存在的丁二烯反应、通过逆反应、去掉hcn之外,通过相似的机制,本文的发明人所相信的是由其实现了lhp失活和丁烯基膦酸二酯形成的反应循环。
图4显示对于配体2来说lhp形成的随时间的时间进程。在含有2500ppm水的顺式-2pn中使用配体2,于75℃维持2天并且之后加入500ppmzncl2,进行研究。如可以看到的,当将路易斯酸zncl2加入至反应环境时,发生了明显的配体(配体2)水解和2,4-二甲苯酚的形成,即lhp形成的副产物的形成。
图5显示在500ppmh2o的存在下在75℃历时5天后,包含二齿亚磷酸酯配体即配体3的镍-亚磷酸酯催化剂的气相色谱法/质谱法痕量。与31pnmr结果一致,在水解的催化剂样品中观察到明显的量的丁烯基和氰基丁基膦酸酯,但是不存在酸性亚磷酸二芳基酯lhp。在不存在所述二齿配体的情况下,观察到酸性lhp产物。
实施例
实施例1-配体1、配体2、和配体3的混合物的水解,其中在混合物中存在由配体3形成的镍亚磷酸酯配合物。
在10ml、厚壁、锥形reacti-vialtm中使用温控reacti-blocktm铝加热块进行实验。使用三角形磁力搅拌棒完成混合。将加热块封闭在氮吹扫箱中。在手套箱内,用在其中也存在配体1和配体2的所述式配体3-镍配合物在戊烯腈中的溶液装入reacti-vialtm中,并且之后一旦在实验开始时转移至加热块即通过微型注射器装入水。初始水浓度是2500ppm,并且将reacti-blocktm维持在75℃。之后以所需间隔移除样品用于借助hplc的分析。在研究结束时,还在所选的样品上得到了31pnmr。对于分析结果数据,参见图6和表1。配体1和配体2是单齿亚磷酸酯配体,配体3是二齿亚磷酸酯配体,并且2,4x是2,4-二甲苯酚。
比较例a.配体1、配体2、和配体3的混合物在不用存在镍催化剂的情况下的水解。
重复实验实施例1,但是删去镍金属的加入,随时间发生配体1、配体2、和配体3的水解和2,4x的累积。数据显示,亚磷酸酯的水解表现出诱导期,接着速率加速,其为自催化反应的特征。对于分析结果数据,参见图7和表1。
比较例b-配体2的水解,其中在混合物中存在由配体4形成的镍亚磷酸酯配合物。
重复实施例1,但是用配体4-镍配合物代替配体3-镍配合物。对于分析结果数据,参见图8和表1。
比较例c-配体1和配体3在戊腈中的混合物的水解,其中在混合物中存在由配体3形成的镍亚磷酸酯配合物。
重复实施例1,但是使用戊腈代替戊烯腈作为溶剂。对于分析结果数据,参见图9和表1。
实施例2-使用由配体3形成的镍配合物来由配体2合成膦酸酯。
用2500ppm水和500ppmzncl2在75℃下过夜将1.2%配体2在pn中的溶液基本水解。借助hplc的反应混合物的分析显示配体2的定量转化,并且gcms分析表明主要形成第一水解产物。之后向反应混合物装入等分部分的配体3-ni配合物,以使溶液中镍的浓度为大约1%,并且之后继续加热5天。在这一时间后,通过gcms和31p-nmr分析溶液,其显示形成了丁烯-和氰基丁基膦酸酯衍生物的配合物混合物。不存在残留的水解产物。对于分析结果数据,参见表1。
比较例d-使用配体5-镍配合物由配体2尝试合成膦酸酯。
重复实施例2,但是使用配体5的镍配合物。仅发现配体2的水解产物。未观察到膦酸酯形成。对于分析结果数据,参见表1。
实施例3-使用由配体3形成的镍配合物来由配体1合成膦酸酯。
重复实施例2,但是用配体1代替配体2。对于分析结果数据,参见表1。
表1:实施例1-3和比较例的结果的总结
在表1中,使用以下缩写。pn:混合的戊烯腈(~87%3pn),vn:戊腈,配体:与镍配位的亚磷酸酯
表1中给出的数据显示,如由实施例2和3所示出的,配体3镍配合物催化由亚磷酸酯的水解产物至膦酸酯的形成,并且如由比较例d所证明的,比配体5更有效。此外,如由实施例1与比较例a的比较所示出的,配体3ni配合物通过抗自催化水解保护由pn催化氢氰化为己二腈得到的反应混合物的亚磷酸酯。此外,如由来自实施例1和比较例b的数据的比较所示出的,表明配体3ni配合物比其他二亚磷酸酯-镍配合物更有优势。此外,如由实施例1和比较例c的数据的比较所示出的,表明在非烯烃溶剂体系如戊腈中,在其中膦酸酯形成是不可能的,却发生自催化亚磷酸酯水解,进一步说明了膦酸酯形成为抗自催化亚磷酸酯水解的保护的方式。
尽管未在这里示出,在与表1中的那些相同但是不存在水的条件下进行的对照实验显示无亚磷酸酯消耗或膦酸酯形成。
本文中引用的所有专利和出版物通过引用结合于此,其结合程度如同具体地并逐一地指出每个单独的出版物是通过引用整体结合的一样。
已经采用的术语和表述用作描述的而且是非限制性的术语,并且并非意在通过使用此类术语和表述排除示出的和描述的特点的任何等价物或其一部分,而是了解到在所要求保护的本发明的范围内可以存在多种修改。因此,应理解的是,尽管已经通过优选的实施特征和任选的特征具体地公开了本发明,可以由本领域技术人员采取在本文中公开的概念的修改和变化,并且此类修改和变化被认为在如由所附权利要求定义的本发明的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!