一种活性维生素D3类药物CD环中间体的制备方法与流程
本发明涉及属于药物化学领域,具体涉及到一种活性维生素d3类药物cd环中间体25-羟基grundmann’s酮的制备方法。
背景技术:
1936年,人们从鳕鱼中发现了维生素d3。以后发现了维生素d3的生理功能是促进肠道钙吸收,诱导骨质钙磷沉着和防止佝偻病。人体中的维生素d3由表皮和皮肤组织中的7-脱氢胆固醇转化而来,或来自于食物。在肝脏羟基化酶的作用下,维生素d3侧链的25位发生羟基化,又在肾脏羟基化酶的作用下,a环的1位羟基化,转化为活性维生素d3(骨化三醇),即1α,25-(oh)2d3,是维生素d3起生理作用的基本形式。维生素d受体不仅存在于小肠、骨胳和肾脏,还存在于人体的各种组织以及各种癌细胞中。活性维生素d3通过与维生素d受体结合,作用于多种靶组织,生理作用也多种多样,不仅能调节体内钙磷代谢,还具有诱导细胞分化、抑制细胞增殖、免疫调节等功能。活性维生素d3类药物骨化三醇、艾尔骨化醇、马沙骨化醇、钙泊三醇、他骨化醇等在临床上用于治疗骨质疏松、肾透析患者继发性甲状旁腺功能亢进症、银屑病和干癣等皮肤病等。活性维生素d3衍生物还有望用于治疗癌症、自身免疫性脑脊髓炎、类风湿性关节炎、系统性红斑狼疮、多发性硬化症、i型糖尿病和炎性肠病等。
25-羟基grundmann’s酮是多种活性维生素d3类药物及活性维生素d3衍生物合成的共通中间体,包括骨化二醇,骨化三醇以及艾尔骨化醇等。虽然已有文献对25-羟基grundmann’s酮的合成方法报道,但在侧链引入、仲羟基保护基脱保护等方面,存在副反应多、总收率低等问题。
技术实现要素:
为了解决现有技术的不足,本发明的目的之一是提供一种活性维生素d3类药物cd环中间体的制备方法,该方法步骤简单、产品收率高,能够大量制备25-羟基grundmann’s酮。
为了实现上述目的,本发明的技术方案为:
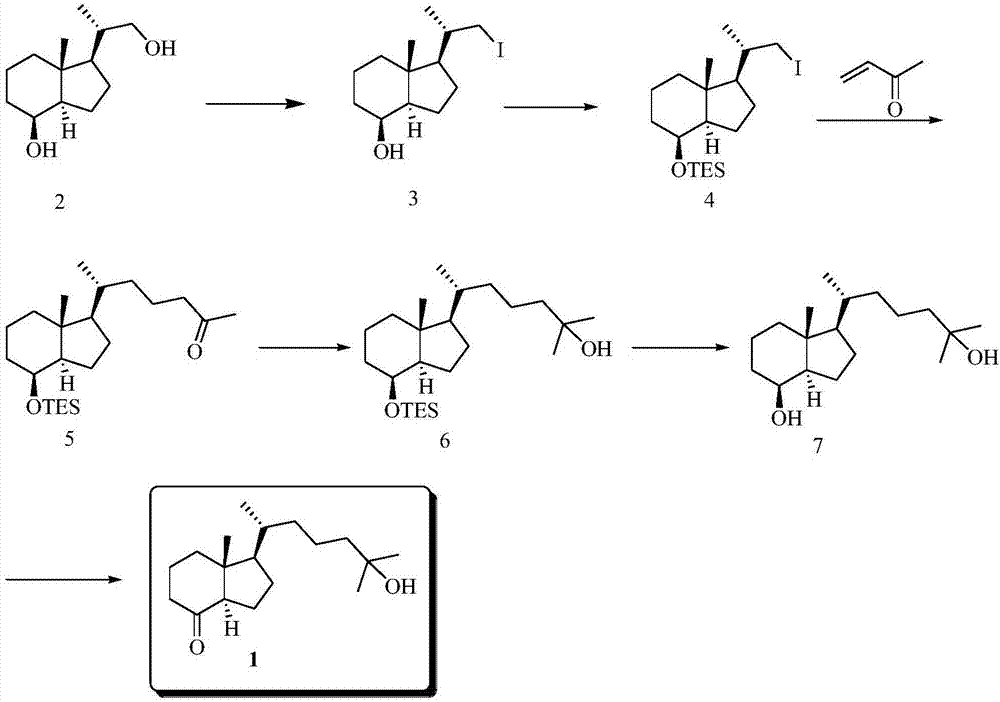
一种活性维生素d3类药物cd环中间体的制备方法,将二醇衍生物2经碘取代获得化合物3,化合物3经过羟基反应得到化合物4,化合物4经过迈克尔加成反应得到化合物5,化合物5经过亲核加成反应得到化合物6,化合物6经过脱保护反应得到化合物7,化合物7经过氧化反应得到化合物1,即活性维生素d3类药物cd环中间体(25-羟基grundmann’s酮)。
首先,本发明通过该合成工艺路线合成了25-羟基grundmann’s酮。其次,本发明的工艺中每个步骤生产的产物的产率较高,使得生产25-羟基grundmann’s酮具有较高的产率。第三,本发明的工艺步骤简单使得副产物较少,且产品收率高,能够适用于工业化大规模生产。
二醇衍生物2的结构式为:
化学物3的结构式为:
化学物4的结构式为:
化学物5的结构式为:
化学物6的结构式为:
化学物7的结构式为:
其合成路线如下:
本发明的目的之二是提供一种上述制备方法在制备活性维生素d3类药物中的应用。
与现有技术相比,本发明的有益效果为:
本发明提出了一种步骤更为简单的制备方法,防止步骤复杂带来的原料损失,从而提高了25-羟基grundmann’s酮的生产效率,降低了生产成本,同时,该方法中每个步骤生产的产物的产率较高,使得生产25-羟基grundmann’s酮具有较高的产率,进一步降低了生产成本。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。
图1为本发明的活性维生素d3类药物cd环中间体的制备方法的合成路线图;
图2为实施例1的合成路线图。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本申请提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本申请所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
本发明中所述的活性维生素d3类药物cd环中间体(25-羟基grundmann’s酮),英文名为25-hydroxygrundmann'sketone,化学名为(1r,3ar,7ar)-octahydro-1-[(1r)-5-hydroxy-1,5-dimethylhexyl]-7a-methyl-4h-inden-4-one,结构式如下:
本发明中所述的二醇衍生物2可以由维生素d2经过氧化后还原获得,也可以由其他方式获得。
正如背景技术所介绍的,现有技术中存在现有合成25-羟基grundmann’s酮存在副反应多、总收率低的不足,为了解决如上的技术问题,本申请提出了一种活性维生素d3类药物cd环中间体的制备方法。
本发明的一种典型的实施方式中,如图1所示,提供了一种活性维生素d3类药物cd环中间体的制备方法,将二醇衍生物2经碘取代获得化合物3,化合物3经过羟基反应得到化合物4,化合物4经过迈克尔加成反应得到化合物5,化合物5经过亲核加成反应得到化合物6,化合物6经过脱保护反应得到化合物7,化合物7经过氧化反应得到化合物1,即活性维生素d3类药物cd环中间体(25-羟基grundmann’s酮)。
二醇衍生物2的结构式为:
化学物3的结构式为:
化学物4的结构式为:
化学物5的结构式为:
化学物6的结构式为:
化学物7的结构式为:
首先,本发明通过该合成工艺路线合成了25-羟基grundmann’s酮。其次,本发明的工艺中每个步骤生产的产物的产率较高,使得生产25-羟基grundmann’s酮具有较高的产率。第三,本发明的工艺步骤简单使得副产物较少,且产品收率高,能够适用于工业化大规模生产。
为了提高化合物3的产率,优选的,向二醇衍生物2的溶液中加入三苯基膦、咪唑及碘,搅拌反应一段时间。
为了得到二醇衍生物2的溶液,进一步优选的,采用四氢呋喃作为溶剂溶解二醇衍生物2。
为了提高原料的反应效率,进一步优选的,二醇衍生物2、三苯基膦、咪唑、碘的摩尔比为1:1.0~1.1:3.0~3.5:1.3~1.5。
为了降低副反应的进行,进一步提升化合物3的收率,进一步优选的,所述搅拌反应时间为7~8h。
为了降低下一步骤的副反应,进一步优选的,对搅拌反应后的溶液进行纯化处理,其步骤为,先加入饱和nahco3溶液进行淬灭反应,再进行萃取,然后对萃取后的有机相进行洗涤干燥,最后进行柱层析获得化合物3。
为了提高化合物4的收率,优选的,向化合物3的溶液中加入碱和氯硅烷,在冰浴下搅拌反应一段时间。
为了得到化合物3的溶液,进一步优选的,采用二氯甲烷作为溶剂溶解化合物3。
为了提高原料的反应效率,进一步优选的,化合物3、碱和氯硅烷的比为1:0.4~0.6:1~1.5,g:g:ml。
进一步优选的,所述碱为2,6-二甲基吡啶或咪唑,所述氯硅烷为叔丁基二甲基氯硅烷、三甲基氯硅烷、三乙基氯硅烷。
为了降低下一步骤的副反应,进一步优选的,对搅拌反应后的溶液进行纯化处理,其步骤为,先进行萃取,然后对萃取后的有机相进行洗涤干燥,最后进行柱层析获得化合物4。
优选的,所述化合物5的制备步骤为,将锌粉和氯化镍加入至吡啶后搅拌均匀,再加入丁烯酮在惰性气体的保护下加热反应一段时间,冷却后获得反应液,然后向所述反应液中逐滴加入化合物4的溶液继续反应一段时间。所述惰性气体为氧化性较弱的气体,如氮气、氦气、氩气等。
进一步优选的,锌粉、氯化镍、丁烯酮的比为5:1:419~420,mol:mol:ml;锌粉与化合物4的摩尔比为1:1.2~1.5。
优选的,向化合物5的溶液中滴加格式试剂,滴加完毕后反应一段时间。
进一步优选的,所述格式试剂为甲基溴化镁。
进一步优选的,滴加完毕后反应12~17h。
进一步优选的,所述格式试剂与化合物5的摩尔比为1:4~4.5。
优选的,制备化合物7所用的脱保护基试剂为四丁基氟化铵(tbaf)、hf/mecn或樟脑磺酸。
优选的,化合物7制备化合物1所用的氧化剂为过硫酸氢钾复合盐(oxone)。
进一步优选的,化合物7与oxone的摩尔比为1:2~3。本发明中化合物7与oxone的摩尔比是指化合物7与oxone中的有效成分过硫酸氢钾(khso5)的摩尔比。
进一步优选的,向化合物7的溶液中加入四丁基溴化铵(bu4nbr)、oxone及四甲基哌啶氮氧化物(tempo),室温下反应一段时间。所述室温为15~30℃。
更进一步优选的,化合物7:tempo:oxone:bu4nbr的摩尔比为1:0.05:2.3:0.1。
本发明还提供一种上述制备方法在制备活性维生素d3类药物中的应用。
为了使得本领域技术人员能够更加清楚地了解本申请的技术方案,以下将结合具体的实施例详细说明本申请的技术方案。
实施例1
二醇衍生物2的合成
将维生素d2(20.0g,50.4mmol)溶于320ml二氯甲烷和80ml甲醇的混合溶液中,通入臭氧,-78℃下搅拌反应3h,停止通入臭氧,通入n2直至蓝色消失。分两次加入nabh4(20.0g,0.528mol),停止制冷,电动搅拌过夜。加入饱和nh4cl溶液、1nhcl溶液淬灭反应。用二氯甲烷萃取多次,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,柱层析(石油醚/乙酸乙酯=5/1),得二醇衍生物2(白色固体状)7.06g,收率71%。
由二醇衍生物2制备获得25-羟基grundmann’s酮的过程,如图2所示,
(1)化合物3的合成
将二醇衍生物2(12.4g,58.4mmol)溶于thf(300ml)中,依次加入三苯基膦(16.12g,61.5mmol),咪唑(12g,176.5mmol),碘(20g,78.8mmol),搅拌反应8h。加入饱和nahco3溶液淬灭反应,用乙酸乙酯萃取2次,有机相分别用饱和硫代硫酸钠溶液、食盐水洗涤,无水硫酸钠干燥。过滤,减压浓缩,柱层析(石油醚/乙酸乙酯=60/1)得化合物3(无色油状物)17g,收率90%。
(2)化合物4的合成
将上步制得的化合物3(15g,46.6mmol),用100ml二氯甲烷溶解,冰浴下搅拌10分钟后加入咪唑(6.66g,97.8mmol),三乙基氯硅烷(15ml),搅拌反应2h。用二氯甲烷萃取3次,饱和食盐水洗涤,无水硫酸钠干燥。过滤,减压浓缩,柱层析(石油醚),得化合物4(无色油状物)20.3g,收率100%。
(3)化合物5的合成
250ml圆底烧瓶中依次加入锌粉(25.85g,0.395mol),nicl2.6h2o(10.25g,79mmol),100ml吡啶,室温下搅拌均匀后,加入丁烯酮(33.15ml),氮气保护下65℃反应2h后,降至25℃。将上步制得的化合物4(23g,52.7mmol)用20ml吡啶溶解后缓慢滴加至上述反应液,搅拌反应7h。用乙酸乙酯萃取,水洗,稀盐酸洗,饱和食盐水洗,无水硫酸钠干燥。过滤,减压浓缩,柱层析(p/e=60:1)得化合物5(无色油状物)13g,收率78.3%。
(4)化合物6的合成
将上步制得的化合物5(13g,34.2mmol)用100ml四氢呋喃溶解后,冰浴下慢慢滴加甲基溴化镁(22.89ml,7.63mmol),搅拌反应17小时。加入饱和氯化铵溶液淬灭反应,用乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。过滤,减压浓缩,得化合物6(粗品)12.4g。
(5)化合物7的合成
将上步制得的化合物6(12.4g,粗品)溶于100ml甲醇中,加入樟脑磺酸(10.9g,46.9mmol),室温下搅拌反应7h。用乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。过滤,减压浓缩,重结晶(p/e=5:1),得化合物7(白色固体)6.552g,两步收率(以化合物5计)68%。
(6)25-羟基grundmann’s酮1的合成
将上步制得的化合物7(6.97g,24.7mmol)用150ml二氯甲烷溶解后,依次加入bu4nbr(798mg,2.48mmol),oxone(19.7g,(含khso58.59g,56.81mmol)),tempo(199.4mg,1.28mmol),室温下搅拌反应5.5h。用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥。过滤,减压浓缩,柱层析(pe/ea=5/1)得化合物1(无色油状)6.5g,收率94%。
二醇衍生物2制备获得25-羟基grundmann’s酮的路线总收率45%。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种丙辛吲哚盐酸盐药物中间体丙辛吲哚的合成方法与制造工艺
- 一种吲哚洛尔药物中间体(一)‑4‑(2‑羟基‑3‑异丙氨基丙氧)吲哚的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 一种氯苯达诺药物中间体邻氯苯甲酸的合成方法与制造工艺
- 一种噢哢类抗癌化合物药物中间体2,4,6‑三羟基苯乙酮的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 一种氯苯达诺药物中间体邻氯苯甲酸的合成方法与制造工艺
- 一种噢哢类抗癌化合物药物中间体2,4,6‑三羟基苯乙酮的合成方法与制造工艺
- 一种萘普生药物中间体1‑(5‑氯‑6‑甲氧基‑2‑奈基)丙酮的合成方法与制造工艺
- 一种萘普生药物中间体6‑甲氧基‑2‑丙酰基萘的合成方法与制造工艺
- 一种噻虫啉药物中间体1,1‑二乙氧基丙烷的合成方法与制造工艺
- 一种阿莫罗芬药物中间体对叔戊基苄基溴的合成方法与制造工艺
- 一种氯胺酮药物中间体环戊烯的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 一种氯苯达诺药物中间体邻氯苯甲酸的合成方法与制造工艺
- 一种噢哢类抗癌化合物药物中间体2,4,6‑三羟基苯乙酮的合成方法与制造工艺
- 一种萘普生药物中间体1‑(5‑氯‑6‑甲氧基‑2‑奈基)丙酮的合成方法与制造工艺
- 一种萘普生药物中间体6‑甲氧基‑2‑丙酰基萘的合成方法与制造工艺
- 一种噻虫啉药物中间体1,1‑二乙氧基丙烷的合成方法与制造工艺
- 一种阿莫罗芬药物中间体对叔戊基苄基溴的合成方法与制造工艺
- 一种氯胺酮药物中间体环戊烯的合成方法与制造工艺
- 一种黄酮类化合物药物中间体1,1‑二苯基‑1‑丙烯的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺
- 一种氯苯达诺药物中间体邻氯苯甲酸的合成方法与制造工艺
- 一种噢哢类抗癌化合物药物中间体2,4,6‑三羟基苯乙酮的合成方法与制造工艺
- 一种萘普生药物中间体1‑(5‑氯‑6‑甲氧基‑2‑奈基)丙酮的合成方法与制造工艺
- 一种萘普生药物中间体6‑甲氧基‑2‑丙酰基萘的合成方法与制造工艺
- 一种阿莫罗芬药物中间体对叔戊基苄基溴的合成方法与制造工艺
- 一种氯胺酮药物中间体环戊烯的合成方法与制造工艺
- 一种黄酮类化合物药物中间体1,1‑二苯基‑1‑丙烯的合成方法与制造工艺
- 一种具有高生物活性的天然活性药物中间体及其制备方法与制造工艺