一种替诺福韦二吡呋酯的制备方法与流程
本发明属于药物化学领域,具体涉及一种用于艾滋病和乙肝治疗的原料药富马酸替诺福韦二吡呋酯关键中间体替诺福韦二吡呋酯的制备方法。
背景技术:
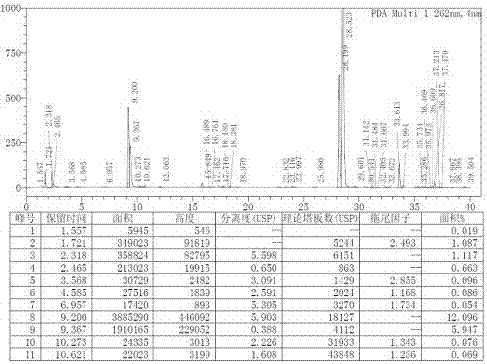
:富马酸替诺福韦二吡呋酯(tenofovirdisoproxilfumarate)是由美国吉利德(gileadsciences)公司研发的新型口服广谱抗病毒药,是目前第一个被fda批准用于治疗hiv-1感染的核苷酸类似物,其重要性也被世界卫生组织承认,并被推荐为一线抗病毒用药。2001年10月该药物获得美国食品药品管理局(fda)批准,用于与其他抗逆转录病毒药物联用治疗hiv-1感染患者;2008年11月,fda又批准该药用于慢性乙型肝炎的治疗。该药于2008年6月被引进中国,批准的适应症是hiv。商品名为富马酸替诺福韦二吡呋酯的化学名为:9-[(r)-2-[[双[[(异丙氧基羰基)氧基]甲氧基]膦酰基]甲氧基]丙基]腺嘌呤富马酸盐(1:1),其结构式为下所示化合物(ii)。富马酸替诺福韦二吡呋酯是一种前药,在体内发生酯基水解转化为替诺福韦而发挥药效。合成富马酸替诺福韦二吡呋酯的关键步骤也是替诺福韦二吡呋酯的合成。目前替诺福韦二吡呋酯的合成方法主要是通过替诺福韦(pmpa)与氯甲基异丙基碳酸酯(cmic)在缚酸剂作用下得到。申请号为201310270413.7的专利,公开了替诺福韦二吡呋酯的制备方法主要是通过替诺福韦与氯甲基异丙基碳酸酯(cmic)在缚酸剂、催化剂聚乙二醇等作用下得到,反应时间长,增加了生产成本。另外,申请号为201610388914.9的专利,公开了通过替诺福韦无水物与氯甲基异丙基碳酸酯(cmic)在缚酸剂、相转移催化剂等作用下得到替诺福韦二吡呋酯,但其相转移催化剂用量大,成本高,且会增加相转移催化剂在产品中的残留,从而影响产品质量。目前,急需要一种成本低、操作简单方法制备替诺福韦二吡呋酯。技术实现要素:通过实验,从提高产品质量以及降低成本角度上考虑,合成替诺福韦二吡呋酯关键在于控制单酯杂质化合物iii(结构如下所示)。为了克服现有技术中的不足,综合考虑工艺的可操作性、经济性和原料药质量,以及适合工业化生产,通过改进反应条件,发现在反应体系中加入特定的催化剂(4-二甲氨基吡啶(dmap))可以大大提高反应产物纯度,减少替诺福韦二吡呋单酯杂质的产生,提高反应转化率;而在相同的反应条件下,其他种类的催化剂却无法达到与之相近似的程度。基于上述原因,本发明实际上提供了:一种替诺福韦二吡呋酯的制备方法,包括以下步骤:1)将替诺福韦(pmpa)、有机碱、催化剂和反应溶剂混合得到混合液;所述催化剂为4-二甲氨基吡啶(dmap);2)在所述混合液中加入氯甲酸异丙基碳酸酯(cmic)反应;3)分离,得到替诺福韦二吡呋酯。本发明所用反应原料替诺福韦(pmpa)为无水化合物。进一步的,步骤1)中,所述有机碱为三乙胺、吡啶或n、n-二异丙基乙胺;优选为三乙胺。进一步的,步骤1)中,所述反应溶剂为n、n-二甲基甲酰胺(dmf)、n、n-二甲基乙酰胺(dmac)或n-甲基吡咯烷酮(nmp);优选为n-甲基吡咯烷酮(nmp)。进一步的,步骤1)中,所述替诺福韦与所述催化剂的摩尔比为1:0.05-0.20。进一步的,步骤1)中,所述替诺福韦与所述反应溶剂质量比为1:3~7。进一步的,步骤1)中,所述替诺福韦与所述氯甲酸异丙基碳酸酯摩尔比为1:3~7。进一步的,步骤1)中,所述替诺福韦与所述有机碱摩尔比为1:2-4。进一步的,步骤2)中,反应温度为常规温度,本发明中采用30~60℃。本发明研究表明,采用本发明催化剂后,反应时间在2-5h,就能够保证较高的纯度和产率。进一步的,步骤3)中,所述分离的步骤为:将步骤2)所得过滤,在得到的滤液中加入水,分液后对水相萃取2-3次,合并有机相后,用饱和碳酸氢钠溶液、氯化钠溶液洗涤,经干燥、过滤、浓缩后析出产物,即为替诺福韦二吡呋酯。进一步的,所述萃取过程中使用的萃取剂为二氯甲烷或乙酸乙酯,优选采用乙酸乙酯。进一步的,所述萃取剂与替诺福韦的质量比为10-20:1。进一步的,滤液中加入的水与替诺福韦的质量比为10-20:1。进一步的,干燥过程中采用干燥剂干燥,所述干燥剂为无水硫酸钠或/和无水硫酸镁,优选采用无水硫酸钠。更进一步的,所述干燥剂与替诺福韦的质量比为0.5-1:1。本发明分离方法,不限于上述方法,也可以为其它现有技术公开能用于分离纯化替诺福韦二吡呋酯的方法。本发明化学反应式为:本发明有益效果:一、本发明选用催化剂4-二甲氨基吡啶(dmap)作为催化剂,无需重结晶等提纯方式即可控制单酯杂质小于4%,工艺相同条件下,采用其它催化剂制得产品中单酯杂质含量为9~14%,无催化剂情况下为15~20%。二、优化反应温度、时间、反应物用量等因素,可增效催化剂作用,对降低单酯杂质含量有重要的意义,通过这些因素还可以提高产品收率达80%以上。三、本发明反应时间短,整个反应过程效率有所提升,且成本也相应降低。附图说明图1为未添加催化剂的反应谱图,单酯rt=7.35min,18.3%;图2为加入催化剂碘化钠的反应谱图,单酯rt=9.2min,12.09%;图3为加入催化剂碘化钾的反应谱图,单酯rt=9.2min,13.40%;图4为加入催化剂聚乙二醇的反应谱图,单酯rt=9.2min,13.62%;图5为加入催化剂聚四乙基溴化铵的反应谱图,单酯rt=9.2min,10.78%;图6为加入催化剂四丁基溴化铵的反应谱图,单酯rt=9.2min,9.04%;图7为实施例1中反应液hplc图,单酯rt=7.39min,2.58%;图8为实施例2中反应液hplc图,单酯rt=7,39min,3.27%;图9为实施例3中反应液hplc图,单酯rt=7.40min,2.31%。具体实施方式以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实例,凡基于本发明上述内容所实现的技术均属于本发明的范围。实施例1:控温10~20℃,将pmpa(287.2g,1.0mol)、三乙胺(202.4g,2.0mol)、4-二甲氨基吡啶(dmap,6.11g,0.05mol)加入到861.0gn-甲基吡咯烷酮溶剂中得到混合液a,搅拌,控温30℃,滴加氯甲基异丙基碳酸酯(457.7g,3.0mol)得到混合液b,然后于30~40℃搅拌反应4小时。反应结束后降至15~25℃,加入乙酸乙酯2.86kg搅拌0.5h后抽滤。滤液加入水2.86kg,分液后水相再加1.43kg乙酸乙酯萃取两次。合并有机相,有机相依次用2.86kg饱和碳酸氢钠溶液,2.86kg饱和氯化钠溶液洗涤,143.0g无水硫酸钠干燥,过滤、浓缩后得到产品。收率85.6%,单酯杂质控制为2.58%。实施例2控温10~20℃,将pmpa(287.2g,1.0mol)、三乙胺(404.8g,4.0mol)、4-二甲氨基吡啶(dmap,24.4g,0.20mol)加入到2.01kgn-甲基吡咯烷酮溶剂中,搅拌,控温25℃滴加氯甲基异丙基碳酸酯(1068.1g,7.0mol),然后于50~60℃搅拌反应2小时。反应结束后降至15~25℃,加入乙酸乙酯5.36kg搅拌0.5h后抽滤。滤液加入纯化水5.32kg,分液后水相再加2.86kg乙酸乙酯萃取三次。合并有机相,有机相依次用5.32kg饱和碳酸氢钠溶液,2.86kg饱和氯化钠溶液洗涤,286.0g无水硫酸钠干燥,过滤、浓缩后得到产品。收率88.3%,单酯杂质控制为3.27%。实施例3控温10~20℃,将pmpa(287.2g,1.0mol)、三乙胺(303.6g,3.0mol)、4-二甲氨基吡啶(dmap,12.2g,0.10mol)加入到1.44kgn-甲基吡咯烷酮溶剂中,搅拌条件下控温不超过30℃滴加氯甲基异丙基碳酸酯(762.3g,5.0mol),然后于40~50℃搅拌反应3小时。反应结束后降至15~25℃,加入乙酸乙酯4.31kg搅拌0.5h后抽滤。滤液加入纯化水4.31kg,分液后水相再加2.15kg乙酸乙酯萃取两次。合并有机相,有机相依次用4.31kg饱和碳酸氢钠溶液,4.31kg饱和氯化钠溶液洗涤,215g无水硫酸钠干燥,过滤、浓缩后得到产品。收率90.6%,单酯杂质控制为2.31%。实施例4根据实施例1操作步骤,对比不同催化剂对单酯杂质含量的影响,结果如下所示:表1不同催化剂对单酯杂质含量的影响催化剂单酯杂质含量——18.3%4-二甲氨基吡啶(dmap)2.6%碘化钠12.09%碘化钾13.40%聚乙二醇13.62%四乙基溴化铵10.78%四丁基溴化铵9.04%注:“——”表示无催化剂添加。尽管这里参照本发明的解释性实施例对本发明进行了描述,但上述实施例仅为本发明较佳的实施方式之一,本发明的实施方式并不受上述实施例的限制,应该理解,本领域技术人员可以设计出很多其他的修改和实施方式,这些修改和实施方式将落在本申请公开的原则范围和精神之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1