水溶性双芴基醌式噻吩衍生物、制备方法及其染色应用与流程
本发明属于精细化工
技术领域:
,涉及到一种双芴基醌式噻吩衍生物、制备方法及其在纤维材料上的染色应用。
背景技术:
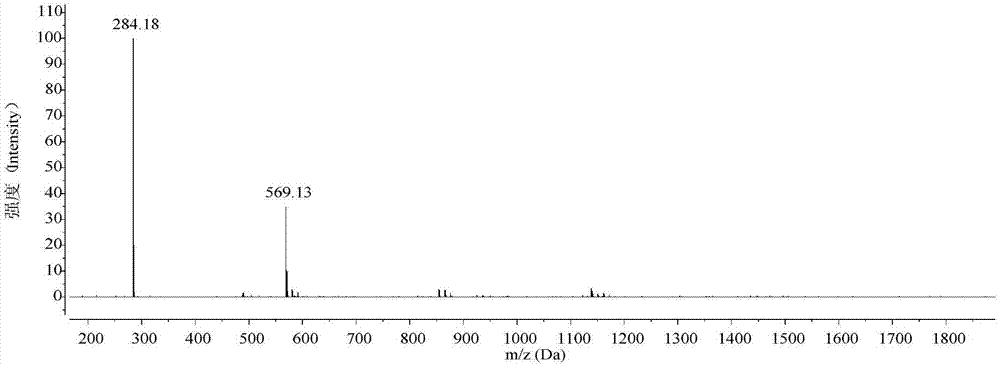
:随着超细涤纶纤维等一批新型纤维的出现及数码喷墨印花技术的普及,染深性问题越来越成为印染行业迫切需要解决的一个难题。通常染深浓颜色的方法是在染色过程中投入大用量染料,不仅会使染色成本大大增加,结果也使染色织物的色牢度降低;即使如此,有些深色织物要求也难以达到。另外,在纺织品数码喷墨印花领域,由于喷印时按需给墨,喷在织物上的墨量很少,染深能力更差,而墨水本身的浓度因为染料溶解度局限而很难增加,即使增加给墨量,也很难得到某些颜色的高色深要求。出现该问题的一个重要原因是当前市售可用染料的摩尔消光系数尚不够高(常见以偶氮、蒽醌为发色母体的染料的摩尔消光系数普遍低于3万l·mol-1cm-1)。醌式结构化合物具有骨架结构刚性强、分子平面性高等特点,该类化合物因其优异的分子间电荷传递能力而作为光电材料被广泛研究(参见:angew.chem.int.ed.2017,56:2250–2259)。共轭大平面醌式结构化合物的另一个特点是其在可见光区域具有极高的摩尔消光系数,选择性吸收能力强。同时,其最大吸收波长和摩尔消光系数均随醌式共轭体系的延长而大幅增加。据报道,醌式结构化合物的摩尔消光系数最高可达20万l·mol-1cm-1(参见:phys.chem.chem.phys.,2015,17:10426–10437)。可见,醌式结构化合物是一类优异的发色体。蒽醌类和三芳甲烷类染料是应用简单醌式结构的两类常见染料。但是其醌式共轭体系较小,摩尔消光系数仍然偏低;且其结构多为苯醌形式,醌式共轭体系难以拓展。双芴基醌式噻吩化合物,目前用于有机光电材料领域,用于制备有机场效应晶体管或有机太阳能电池等。未见有将这类化合物用于染色用染料的报道。技术实现要素:本发明要解决的技术问题是提供一种水溶性双芴基醌式噻吩衍生物、其制备方法和染色应用。为了解决上述技术问题,本发明提供一类双芴基醌式噻吩衍生物,其结构式为如下通式i所示:m为氢、钠;n为1、2、3、4。作为本发明的双芴基醌式噻吩衍生物的改进,其结构式为如下任一:及其相应的钠盐形式:n为1、2、3、4。本发明还同时提供了上述双芴基醌式噻吩衍生物的合成方法,包括以下步骤:以双芴基醌式噻吩化合物(通式ⅱ所示)为原料,经磺化剂通过磺化反应,得到双芴基醌式噻吩衍生物;所述磺化反应温度为-10~100℃,反应时间为0.5~6h(即,以薄层色谱检测原料反应完全为止);磺化剂与双芴基醌式噻吩化合物ii的摩尔比为10:1~500:1;所述磺化剂为浓硫酸(优选)、发烟硫酸、so3、氯磺酸;所述浓硫酸是指质量分数≥70%的硫酸溶液,优选质量分数≥95%的硫酸溶液;所述双芴基醌式噻吩化合物的结构式为如下通式ⅱ所示:反应式如下:说明:双芴基醌式噻吩化合物可参照adv.mater.,2016,28(8),1585-1590和tetrahedronlett.,1991,32(34),4367-4370以及美国专利us20110303909制备而得。作为本发明的双芴基醌式噻吩衍生物的合成方法的改进:当双芴基醌式噻吩衍生物是结构式为i-a(i-a1、i-a2)的化合物时,磺化反应温度为-10~0℃;当双芴基醌式噻吩衍生物是结构式为i-b的化合物时,磺化反应温度为15~25℃;当双芴基醌式噻吩衍生物是结构式为i-c(i-c1、i-c2)的化合物时,磺化反应温度为40~50℃;当双芴基醌式噻吩衍生物是结构式为i-d的化合物时,磺化反应温度为80~100℃。说明:产物i-a是同分异构体i-a1和i-a2的混合物,产物i-c是同分异构体i-c1和i-c2的混合物,均难以分离。由于只是芳环上磺酸基位置发生了改变,供吸电子效应几乎没有变化,因此可以认为同分异构体不会影响混合物的吸收性质,可从下文所测得的紫外-可见吸收光谱中得到证明;混合物不影响作为染料对织物的染色。作为本发明的双芴基醌式噻吩衍生物的合成方法的改进:以相对低级磺化物作为原料,通过磺化反应制备获得相对高级磺化物;磺化反应温度为30~100℃,反应时间为0.5~6h(以薄层色谱检测原料反应完全为止);磺化剂与相对低级磺化物的摩尔比为10:1~500:1;所述相对高级磺化物是指磺化反应中磺酸基数量不少于两个的磺化产物,包括化合物i-b、i-c和i-d;所述相对低级磺化物是指磺化反应中磺酸基数量不多于三个的芴基醌式噻吩衍生物,包括化合物i-a、i-b和i-c;磺化剂为浓硫酸(优选)、发烟硫酸、so3、氯磺酸;浓硫酸是指质量分数≥70%的硫酸溶液,优选质量分数≥95%的硫酸溶液。反应式如下:式中p<q;作为本发明的双芴基醌式噻吩衍生物的合成方法的进一步改进:当高级磺化物为i-b时,磺化反应温度为15~25℃;当高级磺化物为i-c时,磺化反应温度为40~50℃;当高级磺化物为i-d时,磺化反应温度为80~100℃。本发明还同时提供了上述双芴基醌式噻吩衍生物的用途:用于对织物进行染色。作为本发明的双芴基醌式噻吩衍生物的用途的改进:所述织物包括蛋白质纤维、纤维素纤维和合成纤维;所述蛋白质纤维包括羊毛纤维、蚕丝纤维;所述纤维素纤维包括棉纤维、麻纤维;所述合成纤维包括锦纶。本发明旨在开发一种具有高摩尔消光系数的染料化合物,发明人通过对近期有关醌式杂环化合物的研究进行总结分析后发现,双芴基醌式噻吩化合物以芴基为端基、醌式噻吩结构为母体,具有更大的醌式共轭体系,据报道,该化合物在甲苯溶液中的摩尔消光系数可达7.3万l·mol-1cm-1(参见:adv.mater.2016,28:1585–1590)。本发明尝试采用磺化反应引入磺酸基,赋予该化合物水溶性,该水溶性双芴基醌式噻吩衍生物(通式i)同样具有较高的摩尔消光系数。染色应用实践表明,通式i所表示的染料具有广阔的应用前景。与同样具有醌式结构的三芳甲烷类染料(如颜色相近的酸性品红)相比,能够获得更深的颜色。本发明具有如下技术优势:本发明的水溶性双芴基醌式噻吩衍生物合成方便,磺酸基数量较易调控;该类衍生物还具有极高的摩尔消光系数,应用于蛋白质纤维、纤维素纤维和合成纤维织物的染色时,染色织物色光艳丽;与常规染料相比,使用相同用量的染料时,能够获得更高的染色深度。附图说明下面结合附图对本发明的具体实施方式作进一步详细说明。图1是化合物i-a(化合物i-a1和i-a2)的质谱图;图2是化合物i-b的质谱图;图3是化合物i-c(化合物i-c1和i-c2)的质谱图;图4是化合物i-d的质谱图;图5是化合物i-a(化合物i-a1和i-a2)的核磁共振氢谱图;图6是化合物i-b的核磁共振氢谱;从该谱中判断i-b是单一纯物质,不存在异构体。图7是化合物i-d的核磁共振氢谱图;图8是化合物ii、i-a、i-b、i-c和i-d在dmf中的紫外-可见吸收光谱图。具体实施方式为了使本发明的目的和内容更加明晰,下面结合实施例和附图进一步详细说明。这些实施例仅是进一步阐述本发明而非本发明的保护仅限于此。为方便说明,水溶性双芴基醌式噻吩化合物均以游离酸形式表示,但实际形式可能是金属盐,更可能是碱金属盐,尤其是钠盐。下述实施例中所使用的原料ii采用文献(adv.mater.,2016,28(8):1585-1590)或专利(us20110303909)所描述的方法合成,其余所用试剂除特别说明之外,均市售可得。浓硫酸是指质量分数≥70%的硫酸溶液。实施例1:化合物i-a的合成在冰浴条件下(温度控制在-10~0℃之间),向装有化合物ii(2g)的单口烧瓶中,逐滴加入预先降温的浓硫酸(2.6ml,温度为0℃,滴加时间为2min),连续搅拌0.5至1h,当薄层色谱检测原料已经消耗完全,向烧瓶中缓慢加入饱和食盐水(15ml)使产物析出,静置后再经过滤、干燥(50℃干燥至恒重),得到产物i-a(化合物i-a1与化合物i-a2的混合物,红色固体,2.45g,收率:98%)。质谱在m/z489.22处出现了[m-h]-离子峰。实施例2:化合物i-b的合成室温下(温度控制在15~25℃之间),向装有化合物ii(2g)的烧瓶中,逐滴加入浓硫酸(5.2ml,滴加时间为5min),连续搅拌1至3h,当薄层色谱检测原料已经消耗完全,将反应烧瓶置于冰水浴中,加饱和食盐水(15ml)使产物析出,静置后再经过滤、干燥(50℃干燥至恒重),得到化合物i-b(红色固体,2.96g,收率:99%)。质谱在m/z569.13处出现了[m-h]-离子峰;在m/z284.18处出现了[m-2h]2-离子峰。实施例3:化合物i-c的合成在加热条件下(温度为40~50℃的油浴中),向装有化合物ii(2g)的烧瓶中,逐滴加入浓硫酸(7.8ml,滴加时间为10min),连续搅拌2至4h,当薄层色谱检测原料已经消耗完全,将反应烧瓶置于冰水浴中冷却,加饱和食盐水(20ml)使产物析出,静置后过滤,采用dmf-乙醚法除去盐分(将滤饼置于5mldmf中,超声波震荡、搅拌使其完全溶解后,加入10ml乙醚有固体析出,过滤得到滤饼,重复dmf溶解-乙醚析出操作三次),干燥(50℃干燥至恒重)后得到产物i-c(化合物i-c1和化合物i-c2的混合物,红色固体,2.79g,收率:80%)。质谱在m/z649.05处出现了[m-h]-离子峰。实施例4:化合物i-d的合成在加热条件下(温度为80至100℃的油浴中),向装有化合物ii(2g)的烧瓶中,小心逐滴加入浓硫酸(10.4ml,滴加时间为15min),连续搅拌4至6h,当薄层色谱检测原料已经消耗完全,将反应烧瓶置于冰水浴中,加饱和食盐水(20ml)稀释使产物析出,静置后过滤,采用dmf-乙醚法提纯(将滤饼置于5mldmf中,超声波震荡、搅拌使其完全溶解后,加入10ml乙醚有固体析出,过滤得到滤饼,重复dmf溶解-乙醚析出操作三次),干燥(50℃干燥至恒重)后得到化合物i-d(红色固体,3.79g,收率:95%)。质谱在m/z729.10处出现了[m-h]-离子峰。将上述化合物ii、化合物i-a、化合物i-b和化合物i-d溶于dmf中,配制成浓度为2×10-5mol/l的溶液,利用紫外-可见分光光度仪检测溶液的紫外-可见吸收光谱,如图8所示。最大吸收波长和摩尔消光系数如表1所述。酸性品红染料为对照实验。表1酸性品红结构式如下:实施例5:以相对低级磺化物i-b为原料合成相对高级磺化物i-d在加热条件下(温度为80至100℃的油浴中),向装有化合物i-b(1.5g)的烧瓶中,小心逐滴加入浓硫酸(5.2ml,滴加时间为5min),连续搅拌2至3h,当薄层色谱检测原料已经消耗完全,将反应烧瓶置于冰水浴中,加饱和食盐水(20ml)稀释使产物析出,静置后过滤,采用dmf-乙醚法提纯(将滤饼置于5mldmf中,超声波震荡、搅拌使其完全溶解后,加入10ml乙醚有固体析出,过滤得到滤饼,重复dmf溶解-乙醚析出操作三次),干燥(50℃干燥至恒重)后得到化合物i-d(红色固体,1.84g,收率:92%)。实验:采用下表2所示染料进行应用实验。表2编号染料名称组成1a化合物i-a1、化合物i-a22b化合物i-b3c化合物i-c1、化合物i-c24d化合物i-d5e酸性品红实验1、溶解度测试称取一定量的染料于250ml的烧杯中,加入100ml25℃的温水,用机械搅拌器搅拌5min后进行抽滤(抽滤时,压力0.075mpa,5a定性滤纸),观察滤纸残留情况。降低化合物浓度(g/l),直至滤纸上无化合物残留,此时的染料浓度(g/l)即为该染料的溶解度。测试结果记录于下表3中:表3染料名称溶解度(g/l)a80b120c150d150实验2、将染料a、b、c、d和e用于羊毛织物的染色羊毛织物的染色工艺:羊毛织物2g,浴比1:50,染料浓度1%(o.w.f),平平加o用量0.5g/l,冰醋酸调节ph值为4。染浴配制完毕后,升温至40℃,将已经在50℃水中润湿的羊毛织物挤干后投入染浴中开始染色。升温至98℃染色60min。染色结束后取出织物,用冷水洗并烘干。染色结果如下表4:表4abcde上染率/%9699866091布样k/s值283326117.4实验3、将染料a、b、c、d和e用于蚕丝织物的染色蚕丝织物的染色工艺:蚕丝织物2g,浴比1:50,染料浓度1%(o.w.f),平平加o用量0.5g/l,冰醋酸调节ph值为3。染浴配制完毕后,将蚕丝织物投入染浴中开始染色。温度升至90℃染色60min。染色结束后取出织物,冷水冲洗并烘干。染色结果如下表5:表5abcde上染率/%9999938890布样k/s值161815115.5实验4、将染料a、b、c、d和e用于锦纶织物的染色锦纶织物的染色工艺:锦纶织物2g,浴比1:50,染料浓度1%(o.w.f),平平加o用量1g/l,冰醋酸调节ph值为4。染浴配制完毕后,升温至50℃,将锦纶织物投入染浴中开始染色。阶梯升温至98℃染色60min。染色结束后取出织物,冷水冲洗并烘干。染色结果如下表6:表6abcde上染率/%9190827581布样k/s值1516128.66.3实验5、将染料a、b、c、d和e用于棉织物的染色棉织物的染色工艺:棉织物2g,浴比1:50,染料浓度1%(o.w.f),平平加o用量1g/l,氯化钠10g/l。染浴配制完毕后,将棉织物投入染浴中开始染色。温度升至95℃染色60min。染色结束后取出织物,冷水冲洗并烘干。染色结果如下表7:表7abcde上染率/%8082735876布样k/s值1319117.46.4综合上述实验结果可知,染料a~d中,染料b对四类织物染色的上染率和k/s值均最好,其综合性能最佳。与对照组实验结果可以看出,虽然最大吸收波长相似,颜色相近,但双芴基醌式噻吩染料具有相对更高的摩尔消光系数,使用相同用量的染料时能够获得更高的色深值。最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1