蒽双咪唑盐化合物及其制备方法与应用与流程
本发明是在国家自然科学基金(基金号:21572159),天津师范大学青年基金(基金号:52XQ1402)和天津市自然科学基金(基金号:11JCZDJC22000)的资助下进行的。
技术领域
本发明属于有机化学技术领域,涉及通过1,8-二硝基蒽醌、1-正丁基咪唑、六氟磷酸铵作为原料的蒽双咪唑盐化合物的制备,更具体的说是1,8-二[1-正丁基-咪唑乙酰氨基]蒽六氟磷酸盐化合物的制备方法及其在荧光识别性能的研究。
背景技术:
蒽类化合物是一种含有三元环的稠环芳烃,它们是重要的化工原料和中间体,经过多种取代反应可制得蒽的各种衍生物,在染料工业中具有非常重要的意义。针对蒽类化合物,许多的研究者开展了许多探索性的工作。通常在进行预先的实验设计路线过程中,最常见的就是通过引入不同的基团,改变原先的固有特性,使其发挥自身更多优势。比如在蒽类化合物中引入咪唑基,可以将蒽和咪唑的特性结合起来,使用蒽作为荧光团,咪唑作为受体制备成荧光识别体系。由于蒽双咪唑类化合物的荧光性质显著,可以用于对一些客体进行荧光探针方面的研究。随着研究的深入和研究范围的拓展,含咪唑的蒽类化合物作为荧光开关的主体化合物必将在化学学科、生命学科、环境分析和临床医学等领域得到应用。
技术实现要素:
本发明的目的在于提供蒽双咪唑盐化合物及其制备方法。
本发明更进一步涉及了蒽双咪唑六氟磷酸盐化合物在荧光识别领域中的应用。
为完成上述各项发明目的,本发明技术方案如下:
具有下述结构的蒽双咪唑盐化合物:
盐指的是六氟磷酸盐,nBu 指的是正丁基。
本发明所述的蒽双咪唑六氟磷酸盐化合物的制备方法,其特征在于按如下的步骤进行:
(1)将硫化钠与与氢氧化钠的水溶液与1,8-二硝基蒽醌的乙醇溶液混合,在回流下搅拌6小时,得到1,8-二氨基蒽醌;其中硫化钠与1,8-二氨基蒽醌的摩尔比为4.5:1;
(2)将硼氢化钠加入到1,8-二氨基蒽醌和氢氧化钠的异丙醇溶液中,在氮气下回流24小时得到1,8-二氨基蒽,其中1,8-二氨基蒽醌与氢氧化钠的摩尔比为4:1;
(3)将氯乙酰氯滴加到1,8-二氨基蒽和三乙胺的二氯甲烷的溶液中。在室温搅拌过夜,产物过滤,用二氯甲烷洗涤得到1,8-二氯乙酰氨基蒽,其中1,8-二氨基蒽与三乙胺的摩尔比为1:2;
(4)将1,8-二氯乙酰氨基蒽与1-正丁基咪唑以摩尔比为1:3的比例加入到1,4-二氧六环溶液中,加热回流反应5天,洗出深黄色粉末,抽滤,用乙醚洗涤固相产物,得到1,8-二[1-正丁基-咪唑乙酰氨基]蒽氯化物。再将1,8-二[1-正丁基-咪唑乙酰氨基]蒽氯化物与NH4PF6以摩尔比为1:3的比例加入到反应器皿内,用有机溶剂溶解后,在室温温度下搅拌反应3天,析出黄色粉末。抽滤,得到1,8-二[1-正丁基-咪唑乙酰氨基]蒽六氟磷酸盐(2);
本发明所述的制备方法,其中所述的原料为1,8-二硝基蒽醌、硫化钠、干燥的氯化钠、硼氢化钠、三乙胺、甲醇、乙腈、二甲基亚砜、氯乙酰氯、1-正丁基咪唑、六氟磷酸铵;
本发明所述的有机溶剂选自三氯甲烷、二氧六环、甲醇、乙醚、乙腈中的一种或几种的混合物。
一种典型的蒽双咪唑六氟磷酸盐化合物:
盐指的是六氟磷酸盐,nBu 指的是正丁基。
典型的蒽双咪唑六氟磷酸盐的分子式为C32H38N6O2P2F12。
特别加以说明的是蒽双咪唑六氟磷酸盐化合物的单晶数据如下(采用Bruker APEX II CCD衍射仪进行测定):
本发明所述蒽双咪唑六氟磷酸盐化合物晶体的制备方法,其特征在于将蒽双咪唑六氟磷酸盐化合物(2)溶于混合溶剂后放入试管中,在非良性溶剂中扩散令其缓慢结晶得到其淡黄色晶体。
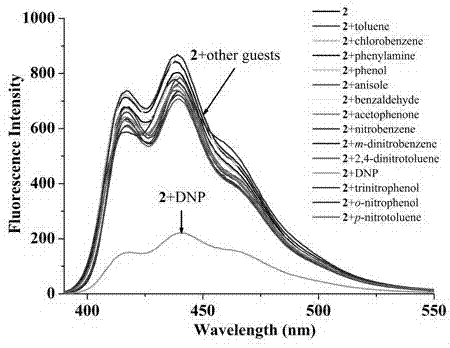
本发明进一步公开了蒽双咪唑六氟磷酸盐化合物在荧光识别领域中的应用;所述的荧光识别指的是对甲苯,氯苯,苯胺,苯酚,苯甲醚,苯甲醛,苯乙酮,硝基苯,间二硝基苯,2,4-二硝基甲苯,苦味酸,邻硝基苯酚,对硝基苯酚,2,4-二硝基苯肼荧光的识别,结果表明:主体2对2,4-二硝基苯肼具有选择性识别能力。
在25˚C下,在蒽双咪唑六氟磷酸盐的乙腈溶液中(主体浓度为:1 × 10-5 mol/L),分别加入相同浓度 (客体浓度为:1 × 10-5 mol/L) 的甲苯,氯苯,苯胺,苯酚,苯甲醚,苯甲醛,苯乙酮,硝基苯,间二硝基苯,2,4-二硝基甲苯,苦味酸,邻硝基苯酚,对硝基苯酚,2,4-二硝基苯肼后测定其荧光光谱,选择吸收峰降低最大的2,4-二硝基苯肼进行滴定。用蒽双咪唑六氟磷酸盐作为主体(主体浓度为:1 × 10-5 mol/L),用微量注射器向其中加入浓度逐渐增大的2,4-二硝基苯肼溶液(0-45× 10-5 mol/L)。主体溶液的激发波长为381nm,发射光谱在390-550 nm有发射峰。每次添加后,8-10分钟达到反应平衡才可记录相应的荧光光谱,使其荧光强度逐渐增强。见附图2和3。
本发明提出的蒽双咪唑六氟磷酸盐化合物是一种在标准状态下可以稳定存在的高级荧光材料,具有制备简洁、荧光感光效果明显的优点,可以用来制作荧光材料和荧光分子识别体系,有望在荧光化学领域得到应用。
附图说明
图1为蒽双咪唑六氟磷酸盐化合物(实施例1)的晶体结构图;
图2为蒽双咪唑六氟磷酸盐化合物(实施例1)在25˚C下,乙腈溶液中(主体浓度为:1 × 10-5 mol/L)分别加入相同浓度 (客体浓度为:1 × 10-5 mol/L) 的甲苯,氯苯,苯胺,苯酚,苯甲醚,苯甲醛,苯乙酮,硝基苯,间二硝基苯,2,4-二硝基甲苯,苦味酸,邻硝基苯酚,对硝基苯酚,2,4-二硝基苯肼后测定其荧光光谱,结果表明:主体2对2,4-二硝基苯肼具有选择性识别能力;
图3为蒽双咪唑六氟磷酸盐化合物(实施例1)在25˚C下,腈溶液中(主体浓度为:1 × 10-5 mol/L)加入不同浓度的2,4-二硝基苯肼溶液(0-45× 10-5 mol/L)后的荧光滴定光谱图;结果表明随着2,4-二硝基苯肼浓度的增加主体的荧光逐渐减弱,当2,4-二硝基苯肼浓度达到一定数值后荧光不再有明显的变化。
具体实施方式
下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。本发明所用原料及试剂均有市售;其中1,8-二硝基蒽醌、硫化钠、干燥的氯化钠、硼氢化钠、三乙胺、甲醇、乙腈、二甲基亚砜、氯乙酰氯、1-正丁基咪唑、六氟磷酸铵等均可以从市场上买到或容易地通过已知的方法制得。
制备本发明化合物所用到的试剂全部来源于天津市科锐思化工有限公司,级别为分析纯。另外需要加以说明的是:所有的实验操作运用Schlenk技术,溶剂经过标准流程纯化。所有用于合成和分析的试剂都是分析纯,并没有经过进一步的处理。熔点通过Boetius区截机测定。1H 和 13C{1H}NRM谱通过汞变量Vx400分光光度计记录,测量区间:400 MHz and 100 MHz。化学位移,δ,参考国际标准的TMS测定。荧光光谱通过Cary Eclipse荧光分光光度计测定。
实施例1
1,8-二氨基蒽醌的制备:
将硫化钠(10.808 g, 45.0 mmol),氢氧化钠(4.280 g, 107.0 mmol)溶于190 mL水中,搅拌使其溶解得到还原溶液。向500 mL三口瓶中加入1,8-二硝基蒽醌(2.980 g, 10.0 mmol),112 mL无水乙醇,搅拌并缓慢升温至乙醇回流。将还原溶液加入其中,回流搅拌6 h。冷却至室温,放入冰箱静置过夜,有大量紫红色沉淀析出。抽滤得到紫红色针状晶体,即产物1,8-二氨基蒽醌。产率:2.320 g (97.5%),熔点:274-275˚C。
1,8-二氨基蒽的制备:
将1,8-二氨基蒽醌(2.000 g, 8.4 mmol)溶解在100 mL异丙醇中,向其中加入氢氧化钠(0.073 g, 1.8 mmol)的水溶液。室温下,通N2对反应体系进行惰性气体保护,向反应体系中分批加入的硼氢化钠(4.000 g, 106.0 mmol),加完后升温至反应体系回流,反应过夜。反应完毕后,冷却至室温,将混合物倒入到250 mL冰水中,析出荧光绿色絮状沉淀。抽滤,充分水洗,干燥,得到黄绿色固体,即为产物1,8-二氨基蒽。产率:1.680 g (96%),熔点:175-176˚C。1H NMR (400 MHZ, DMSO-d6): δ 5.870 (s, 4H, NH2), 6.55 (t, J = 4.1 Hz, 2H, AnH), 7.19 (d, J = 4.1 Hz, 4H, AnH), 8.17 (s, 1H, AnH), 8.80 (s, 1H, AnH).
1,8-二氯乙酰氨基蒽的制备:
将1,8-二氨基蒽(2.000 g,9.6 mmol)溶解在150ml无水二氯甲烷中,向其中加入三乙胺(2.332 g,23.1 mmol),冰盐浴下搅拌溶解。再将氯乙酰氯(2.604 g,23.1 mmol)用8mL无水二氯甲烷稀释并装入恒压滴液漏斗,冰盐浴下向1,8-二氨基蒽的二氯甲烷溶液中滴加氯乙酰氯的二氯甲烷溶液,滴加完毕后恢复室温,搅拌过夜,产物以灰绿色的沉淀析出。待反应完全后抽滤,二氯甲烷多次淋洗,得到灰绿色固体,即为1,8-二氯乙酰氨基蒽。产率: 3.400 g (98.3%), 熔点: > 320 ˚C。 1H NMR (400 MHZ, DMSO-d6): δ 4.55 (s, 4H, CH2), 7.56 (q, J = 5.2 Hz, 2H, AnH), 7.72 (d, J = 7.1 Hz, 2H, AnH), 8.01 (d, J = 8.5 Hz, 2H, AnH), 8.68 (s, 1H, AnH), 8.87 (s, 1H, AnH), 10.53 (s, 2H, NH).
1,8-二[1-正丁基-咪唑乙酰氨基]蒽氯化物的制备(1):
将1,8-二氯乙酰氨基蒽(1.000 g,2.8 mmol)溶于1,4-二氧六环 (100 mL)溶液中,加热搅30min。然后加入1-正丁基咪唑(0.799 g,8.3 mmol),回流搅拌5天,析出深黄色粉末,产物过滤,用1,4-二氧六环洗涤,得到1,8-二[1-正丁基-咪唑乙酰氨基]蒽氯化物。产率:1.400 g (83.3%),熔点:276-280˚C。1H NMR (400 MHz, DMSO-d6): δ 0.92 (t, J = 7.4 Hz, 6H, CH3), 1.29 (q, J = 15.4 Hz, 4H, CH2), 1.83 (q, J = 7.2 Hz, 4H, CH2), 4.27 (t, J = 7.1 Hz, 4H, CH2), 5.80 (s, 4H, CH2), 7.54 (t, J = 7.9 Hz, 2H, AnH), 7.87 (s, 2H, ArH), 7.98 (t, J = 9.0 Hz, 4H, ArH), 8.04 (d, J = 7.3 Hz, 2H, AnH), 8.65 (s, 1H, AnH), 9.43 (s, 2H, imiH), 9.91 (s, 1H, AnH), 11.34 (s, 2H, NH) (imi = imidazole)。
1,8-二[1-正丁基-咪唑乙酰氨基]蒽六氟磷酸盐的制备(2):
将1,8-二[1-正丁基-咪唑乙酰氨基]蒽氯化物(1.000 g,1.8 mmol)溶于甲醇(100 mL),向反应体系中加入的六氟磷酸铵(0.884 g,5.4 mmol),立即形成深黄色沉淀。过滤收集黄色粉末,用少量甲醇洗涤,真空下干燥,得到1,8-二[1-正丁基-咪唑乙酰氨基]蒽六氟磷酸盐。产率:1.000 g (74%). 熔点:194-198 ˚C。 1H NMR (400 MHz, DMSO-d6): δ 0.93 (t, J = 7.3 Hz, 6H, CH3), 1.28 (m, 4H, CH2), 1.87 (m, 4H, CH2), 4.27 (t, J = 6.8 Hz, 4H, CH2), 5.62 (s, 4H, CH2), 7.57 (t, J = 7.9 Hz, 2H, AnH), 7.90 (m, 6H, ArH), 7.99 (d, J = 8.5 Hz, 2H, AnH), 8.68 (s, 1H, AnH), 9.33 (s, 2H, imiH), 9.39 (s, 1H, AnH), 11.02 (s, 2H, NH) (imi = imidazole). 13C NMR (100 MHz, DMSO-d6): δ 164.77 (C=O), 139.23 (imi NCN), 137.48, 137.41, 132.96, 131.65, 127.08, 125.55, 125.38, 125.20, 124.11, 124.00, 121.91, 119.48, 116.94 (ArC), 64.85, 51.73 (CH2), 48.71, 48.79 (imi NCH2), 31.34 (CH2), 18.74 (CH2), 13.22 (CH3), (imi = imidazole)。.
盐指的是六氟磷酸盐,nBu 指的是正丁基。
晶体结构见说明书附图1:
实施例1
蒽双咪唑六氟磷酸盐化合物的晶体,其晶体结构参数如下:
晶体数据和结构精修参数包含在支持性信息中。在Bruker APEX II CCD衍射仪上进行,实验温度为293(2)K, 在50kV 和20mA下,用Mo-Ka 辐射(0.71073Å)操作,用SMART和SAINT软件进行数据收集和还原,q 的范围是 1.8 < q < 25º。应用SADABS 程序进行经验吸收矫正。晶体结构由直接方法解出,用SHELXTL包对全部非氢原子坐标各向异性热参数进行全矩阵最小二乘法修正。
应用实例1
25˚C下,在蒽双咪唑六氟磷酸盐化合物2的乙腈中(浓度为1 × 10-5 mol/L),分别加入相同浓度(浓度为1 × 10-5 mol/L)的甲苯,氯苯,苯胺,苯酚,苯甲醚,苯甲醛,苯乙酮,硝基苯,间二硝基苯,2,4-二硝基甲苯,苦味酸,邻硝基苯酚,对硝基苯酚,2,4-二硝基苯肼后测定其荧光光谱,见附图2,从附图2中可以看出主体2对2,4-二硝基苯肼具有选择性识别能力;
荧光滴定通过Cary Eclipse荧光分光光度计用1cm路径长的石英槽测定的。滴定的进行是将主体2 (浓度为1 × 10-5 mol/L)放入4 mL的比色皿中,并用微量注射器加入浓度逐渐增大的2,4-二硝基苯肼溶液(0-45× 10-5 mol/L)。主体溶液的激发波长为383nm,发射光谱在390-550nm有发射峰。每次添加后,8-10分钟达到反应平衡才有吸收的荧光强度,数据分析使用Origin 8.0,见附图3,从附图3中可以看出:随着2,4-二硝基苯肼浓度的增加主体的荧光逐渐减弱,当2,4-二硝基苯肼浓度达到一定数值后荧光不再有明显的变化。
综上所述,本发明的内容并不局限在实例中,相同领域内的有识之士可以在本发明的技术指导思想之内可以轻易提出其他的实例,但这种实例都包括在本发明的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!