一种基因敲除选育bai2基因缺失型斑马鱼的方法与流程
本发明涉及基因敲除领域,特别是公开了一种基因敲除选育bai2基因缺失型斑马鱼的方法。
背景技术:
bai2(又称adgrb2,adhesiongprotein-coupledreceptorb2)基因位于斑马鱼第19号染色体上,包含14个外显子,cdna全长1904bp,同时通过基因差异表达谱分析和基因组关联分析等,发现bai2基因在人类胚胎早期的多个组织中都有表达,特别的是,该基因与斑马鱼左右不对称发育密切相关。
斑马鱼与人类心脏发育过程中的基因、信号通路有高度同源性,且bai2基因进化上较为保守,研究发现bai2在斑马鱼胚胎早期表达量较高。而且,与其他动物模型相比,斑马鱼具有个体小、易于饲养、发育快、繁殖能力强、体外受精、胚胎体外发育且透明等优点。
基因打靶技术起源于20世纪80年代末,是一种通过对基因组进行定点修饰来研究基因功能的重要方法手段,也可用于治疗人类的各种遗传性疾病。该技术主要是利用缺失突变、基因灭活、染色体大片段删除以及外源基因导入等方式来改变生物的遗传信息,并且在生殖系中稳定遗传后表达突变性状,从而研究生物体内特定基因在生长发育过程中的作用,所以这类技术手段已成为现代分子生物学研究热点。传统的基因打靶技术是建立在胚胎干细胞(esc)和同源重组技术的基础之上,故打靶技术效率极低。2013年初,一种全新的人工核酸内切酶clusteredregularlyinterspacedshortpalindromicrepeats(crispr)/crispr-associated(cas)9,该技术具有操作简单、成本低的特点,能更高效、更精确地在生物体基因组中沉默特定基因,且可同时对靶基因上多个位点进行剪切,沉默任意数目的单个基因,但与此同时,该技术存在一定的缺陷,其脱靶率相对较高。
技术实现要素:
针对现有基因打靶技术中存在的脱靶率相对较高问题,本发明提供了一种基因敲除选育bai2基因缺失型斑马鱼的方法,能更高效且更精确地在生物体基因组中沉默特定基因,且制作简单、成本低,还可同时对靶基因上多个位点进行剪切,沉默任意数目的单个基因,脱靶率较低。与此同时,通过研究bai2基因的缺失与其他器官的发育的相关性,这将为医学研究提供更多信息
解决上述技术问题的技术方案如下:
1)分别设计crispr/cas9基因敲除靶位点和检测引物
在nationalcenterforbiotechnologyinformation(ncbi)上查询斑马鱼bai2基因的基因组dna序列,在网站smart(http://smart.embl-heidelberg.de/)上分析其功能结构域,根据crispr/cas敲除原理,在网站thezifittargeter(http://zifit.partners.org/zifit/)上设计bai2基因的靶位点;
两对特异性靶位点pcr引物如下:
f1(靶位点a正向引物):
tgtaatacgactcactataggtacggctccttctcgctcgttttagagctagaaatagc
f3(靶位点b正向引物):
tgtaatacgactcactataggggaccatacagagttccagttttagagctagaaatagc
r(共用的反向引物):aagcaccgactcggtgccact
pcr检测引物:
f(5’-tgtctgtgctgtcatccctttt-3’)
r(5’-aagcattcctcaaaccaccggc-3’)
2)构建grna表达载体以及特异性grna体外合成
a、首先将grna骨架克隆到p42250载体上;
b、特异性grna体外合成
用bsai限制性内切酶线性化此质粒;酶切反应总体积为20μl,体系如下:
p42250载体2μl
10×buffer2μl
bsai限制性内切酶1.5μl
ddh2o14.5μl
总体积20μl
混匀后于37℃水浴,酶切4h以上;
c、以线性化的p42250载体为模板,通过下面特异性引物进行pcr,扩增出用于特异性grna合成的双链dna;
正向特异性靶位点引物f1或f3:t7启动子20bp+靶序列20bp+sgrna上游骨架,
反向引物r:20bpsgrna下游骨架,
pcr反应体系如下:
ddh2o18μl
2×buffer25μl
dntps(10mm)1μl
primerf1(或f3,10um)2μl
primerr(10um)2μl
模板(p42250载体)1μl
ex-taqdna聚合酶1μl
总体积50μl
震荡混匀之后,4℃离心,于pcr仪上进行扩增反应,待反应结束后,取1μl样品点样于2.0%琼脂糖凝胶,进行电泳检测,凝胶成像系统拍摄结果;
d、检测确定条带正确之后,进行琼脂糖凝胶dna回收,纯化回收pcr产物;
e、测定纯化的dna浓度,再以此dna为模板,用20μl体系进行体外转录,合成特异性grna;转录实验中所用tip头,ep管均为depc处理过的rnase-free产品,具体操作如下:
体外转录反应体系:
模板dna12μl
10×buffer2μl
ratp(10mm)1μl
rutp(10mm)1μl
rctp(10mm)1μl
rgtp(10mm)1μl
t7rna聚合酶2μl
总体积20μl
将反应物全部加入1.5mlrnase-free的ep管中,混匀之后,于37℃水浴2.5h;然后向转录体系中加入1μldna酶,放置于37℃水浴锅中反应30min,用以消化dna模板;再取1μl转录终产物,即grna进行琼脂糖凝胶电泳,以检测转录效率;
f、特异性grna的纯化
用rneasyminikit试剂盒纯化转录成功的grna,保存于-20℃;吸取纯化后的grna溶液1μl进行浓度检测
3)斑马鱼胚胎的显微注射
在受精后30min之内,用吸管吸取胚胎转移至用琼脂糖制作的显微注射专用培养皿中,在进行显微注射之前,将cas9蛋白和grna配成混合液,充分混匀,其中,cas9蛋白的浓度为5μg/μl,grna的终浓度在30-40ng/μl之间,注射约1.8nlcas9蛋白和grna混合液于一细胞期的受精卵内;注射过的受精卵放置于e3水中,28℃孵化;在体式显微镜下观察胚胎表型,筛选正常发育的胚胎用于靶位点突变分析;
显微注射体系如下:
cas9蛋白5μg/μl
grna30-40ng/μl
nuclease-freewater
total3μl;
4)sanger测序检测靶位点的有效性
对斑马鱼胚胎进行显微注射之后,挑选部分发育正常的早期胚胎,检测其bai2基因是否存在突变,可以提前确认此次选择的靶位点是否有效果,显微注射操作是否规范;
a、提取斑马鱼基因组
(1)斑马鱼胚胎受精50小时后(50hpf),分别收集野生型(做对照)和注射后胚胎于1.5mlep管中,每管5颗胚胎,按照下述方法提取基因组dna,具体步骤如下:
(2)向装有胚胎的ep管中加入100μl细胞裂解液,1μl蛋白酶k,放置于55℃恒温箱中裂解过夜;
(3)裂解完成后,放在振荡器上充分震荡,加入等体积预先冷却的异丙醇于ep管中,充分颠倒混匀,于4℃条件下,12000rpm离心10min,倒掉上清液;
(4)加入75%乙醇200μl,于4℃条件下,12000rpm离心5min,弃上清液,室温风干8-10min;
(5)加入10μl去离子水,充分吹打混匀,即得斑马鱼基因组dna;
b、pcr扩增目的序列
提取基因组dna后,根据crispr靶位点上下游约100-250bp的基因组区域,利用primer3.0软件设计引物序列以扩增出目的dna片段;
pcr反应体系如下:
2×estaqmastermix10μl
primerf(10μm)0.6μl
primerr(10μm)0.6μl
模板(基因组dna)1μl
ddh2o7.8μl
总体积20μl
震荡混匀之后,4℃离心,于pcr仪上进行扩增反应;
c、用1.8%琼脂糖凝胶电泳检测pcr产物,
d、送部分pcr产物进行sanger测序,由测序的峰图来初步获得插入或缺失的信息;
e、注射两个月之后,进行剪尾鉴定,同上鉴定步骤;
5)获得可遗传的斑马鱼突变体的f1代
通过前面一系列筛选确定了斑马鱼突变体f0代,紧接着将f0代突变体分别与野生型斑马鱼杂交得到f1代胚胎,置于28℃培养,在初期观察f1代的存活率;受精两天后,每个突变体f1代分别取5个胚胎进行突变遗传性鉴定;将每个胚胎单独提取基因组,然后pcr扩增出571bp的靶位点附近区域,观察pcr扩增是否会出现小带;如果此突变可以遗传到f1代,则pcr扩增会出现小于571bp的小带;如果从f1代胚胎中检测到存在突变,则将斑马鱼突变体的f1代养大至1-2个月;再分别对每条f1代斑马鱼成鱼进行剪尾,筛选f1代突变体(具体方法如前面所述)。
本发明的有益效果是:
通过crispr/cas9基因编辑技术,在斑马鱼的bai2基因上设计合适的打靶位点,在体外合成的特异性grna和cas9蛋白,斑马鱼胚胎进行显微注射,sanger测序检测靶位点的有效性;
本发明能更高效且更精确地在生物体基因组中沉默特定基因,制作简单、成本低,还可同时对靶基因上多个位点进行剪切,以达到沉默任意数目的单个基因的目的。且干扰掉bai2基因,并且通过遗传学手段研究其功能,有助于进一步揭示心脏形态发生的整个过程以及调控这些过程的分子机制,在医学上心脏疾病病理的理解和新的治疗方案的研发中具有十分重要的意义。
附图说明
图1为crispr/cas9打靶系统的原理图;
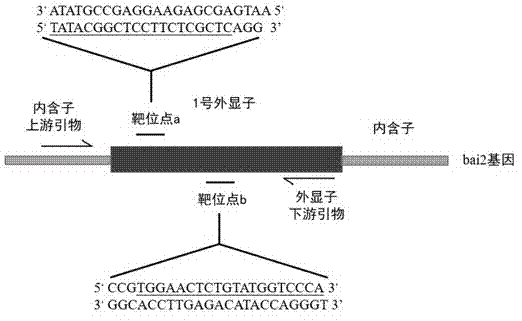
图2为bai2基因上打靶位点结构图;
图3为斑马鱼f1代电泳结果图;
图4为缺失型和wt型基因序列正向对比;
图5为靶位点附近缺失对比。
具体实施方式
下面结合说明书附图和具体的实施例,对本发明作详细描述
实施例1:
1)分别设计crispr/cas9基因敲除靶位点和检测引物
在nationalcenterforbiotechnologyinformation(ncbi)上查询斑马鱼bai2基因的基因组dna序列,在网站smart(http://smart.embl-heidelberg.de/)上分析其功能结构域,根据crispr/cas敲除原理,在网站thezifittargeter(http://zifit.partners.org/zifit/)上设计bai2基因的靶位点;
两对特异性靶位点pcr引物如下:
f1(靶位点a正向引物):
tgtaatacgactcactataggtacggctccttctcgctcgttttagagctagaaatagc
f3(靶位点b正向引物):
tgtaatacgactcactataggggaccatacagagttccagttttagagctagaaatagc
r(共用的反向引物):aagcaccgactcggtgccact
pcr检测引物:
f(5’-tgtctgtgctgtcatccctttt-3’)
r(5’-aagcattcctcaaaccaccggc-3’)
2)构建grna表达载体以及特异性grna体外合成
a、首先将grna骨架克隆到p42250载体上;
b、特异性grna体外合成
用bsai限制性内切酶线性化此质粒;酶切反应总体积为20μl,体系如下:
p42250载体2μl
10×buffer2μl
bsai限制性内切酶1.5μl
ddh2o14.5μl
总体积20μl
混匀后于37℃水浴,酶切4h以上;
c、以线性化的p42250载体为模板,通过下面特异性引物进行pcr,扩增出用于特异性grna合成的双链dna;
正向特异性靶位点引物f1或f3:t7启动子20bp+靶序列20bp+sgrna上游骨架,
反向引物r:20bpsgrna下游骨架,
pcr反应体系如下:
ddh2o18μl
2×buffer25μl
dntps(10mm)1μl
primerf1(或f3,10um)2μl
primerr(10um)2μl
模板(p42250载体)1μl
ex-taqdna聚合酶1μl
总体积50μl
震荡混匀之后,4℃离心,于pcr仪上进行扩增反应,待反应结束后,取1μl样品点样于2.0%琼脂糖凝胶,进行电泳检测,凝胶成像系统拍摄结果;
d、检测确定条带正确之后,进行琼脂糖凝胶dna回收,纯化回收pcr产物;
e、测定纯化的dna浓度,再以此dna为模板,用20μl体系进行体外转录,合成特异性grna;转录实验中所用tip头,ep管均为depc处理过的rnase-free产品,具体操作如下:
体外转录反应体系:
模板dna12μl
10×buffer2μl
ratp(10mm)1μl
rutp(10mm)1μl
rctp(10mm)1μl
rgtp(10mm)1μl
t7rna聚合酶2μl
总体积20μl
将反应物全部加入1.5mlrnase-free的ep管中,混匀之后,于37℃水浴2.5h;然后向转录体系中加入1μldna酶,放置于37℃水浴锅中反应30min,用以消化dna模板;再取1μl转录终产物,即grna进行琼脂糖凝胶电泳,以检测转录效率;
f、特异性grna的纯化
用rneasyminikit试剂盒纯化转录成功的grna,保存于-20℃;吸取纯化后的grna溶液1μl进行浓度检测
3)斑马鱼胚胎的显微注射
在受精后30min之内,用吸管吸取胚胎转移至用琼脂糖制作的显微注射专用培养皿中,在进行显微注射之前,将cas9蛋白和grna配成混合液充分混匀,其中,cas9蛋白的浓度为5μg/μl,grna的终浓度在30-40ng/μl之间,注射约1.8nlcas9蛋白和grna混合液于一细胞期的受精卵内;注射过的受精卵放置于e3水中,28℃孵化;在体式显微镜下观察胚胎表型,筛选正常发育的胚胎用于靶位点突变分析;
显微注射体系如下:
cas9蛋白5μg/μl
grna30-40ng/μl
nuclease-freewater
total3μl;
4)sanger测序检测靶位点的有效性
对斑马鱼胚胎进行显微注射之后,挑选部分发育正常的早期胚胎,检测其bai2基因是否存在突变,可以提前确认此次选择的靶位点是否有效果,显微注射操作是否规范;
a、提取斑马鱼基因组
(1)斑马鱼胚胎受精50小时后(50hpf),分别收集野生型(做对照)和注射后胚胎于1.5mlep管中,每管5颗胚胎,按照下述方法提取基因组dna,具体步骤如下:
(2)向装有胚胎的ep管中加入100μl细胞裂解液,1μl蛋白酶k,放置于55℃恒温箱中裂解过夜;
(3)裂解完成后,放在振荡器上充分震荡,加入等体积预先冷却的异丙醇于ep管中,充分颠倒混匀,于4℃条件下,12000rpm离心10min,倒掉上清液;
(4)加入75%乙醇200μl,于4℃条件下,12000rpm离心5min,弃上清液,室温风干8-10min;
(5)加入10μl去离子水,充分吹打混匀,即得斑马鱼基因组dna;
b、pcr扩增目的序列
提取基因组dna后,根据crispr靶位点上下游约100-250bp的基因组区域,利用primer3.0软件设计引物序列以扩增出目的dna片段;
pcr反应体系如下:
2×estaqmastermix10μl
primerf(10μm)0.6μl
primerr(10μm)0.6μl
模板(基因组dna)1μl
ddh2o7.8μl
总体积20μl
震荡混匀之后,4℃离心,于pcr仪上进行扩增反应;
c、用1.8%琼脂糖凝胶电泳检测pcr产物,
d、送部分pcr产物进行sanger测序,由测序的峰图来初步获得插入或缺失的信息;
e、注射两个月之后,进行剪尾鉴定,同上鉴定步骤;
5)获得可遗传的斑马鱼突变体的f1代
通过前面一系列筛选确定了斑马鱼突变体f0代,紧接着将f0代突变体分别与野生型斑马鱼杂交得到f1代胚胎,置于28℃培养,在初期观察f1代的存活率;受精两天后,每个突变体f1代分别取5个胚胎进行突变遗传性鉴定;将每个胚胎单独提取基因组,然后pcr扩增出571bp的靶位点附近区域,观察pcr扩增是否会出现小带;如果此突变可以遗传到f1代,则pcr扩增会出现小于571bp的小带;如果从f1代胚胎中检测到存在突变,则将斑马鱼突变体的f1代养大至1-2个月;再分别对每条f1代斑马鱼成鱼进行剪尾,筛选f1代突变体(具体方法如前面所述)
图1为crispr/cas9打靶系统的原理图;图2为bai2基因上打靶位点的结构图;图3为wt型和缺失型f1代成鱼pcr电泳图,将测序结果与野生型序列(571bp)进行对比,发现bai2两个靶位点都有碱基缺失,并且从靶位点a处开始到靶位点b处,共有连续的227个碱基的缺失;图4为缺失型和wt型基因序列正向对比图;图5为靶位点附近缺失对比,由于筛选到的突变体f1代的bai2基因部分碱基缺失造成整个基因的移码突变,这会严重影响斑马鱼bai2基因的表达,从而对斑马鱼心脏的发育造成了一定影响。
- 还没有人留言评论。精彩留言会获得点赞!