以超强碱法制备1,2,3,4-四氢-1-萘甲酸的方法与流程
本发明涉及化合物制备技术领域,更具体涉及一种一步法制备1,2,3,4-四氢-1-萘甲酸的方法。
背景技术:
盐酸帕洛诺司琼,商品名阿乐喜,由瑞士helsinnhealthcaresa公司研制,是第二代5-ht3受体拮抗剂,盐酸帕洛诺司琼于2003年7月获得fda审批通过,在美国、日本等极少数发达国家实现了上市。为亲和力较强的5-ht3受体选择性拮抗剂,5-ht3受体位于延髓最后区的催吐化疗感受区中央和周围的迷走神经末梢,化疗药物通过刺激小肠嗜铬细胞释放5-ht,5-ht再激活迷走传入神经的5-ht3受体,产生呕吐反射,盐酸帕洛诺司琼通过抑制5-ht3受体,从而达到止吐效果。
1,2,3,4-四氢-1-萘甲酸是盐酸帕洛诺司琼重要中间体,进一步手性拆分得到s-1,2,3,4-四氢-1-萘甲酸,虽然文献报道了很多手性制备s-1,2,3,4-四氢-1-萘甲酸的方法,但目前能工业化的仍然是拆分消旋体1,2,3,4-四氢-1-萘甲酸得到。因此高效制备1,2,3,4-四氢-1-萘甲酸具有重要商业价值。
目前报道的1,2,3,4-四氢-1-萘甲酸的合成方法主要有以下几种:
方法一:
方法二:
方法三:
方法一,起始原料虽然便宜,但步骤长,用到二氯亚砜设备腐蚀大,最致命的是用到氰化钠上氰基,氰化钠剧毒,使用受到了严格限制,虽然我们也尝试了其他氰化方法,要么收率低,要么不反应,并且步骤长收率低,这条路线不具备工业化价值。
方法二,原料便宜易得,但用到高压氢化,雷尼镍具有巨大的安全风险,我们在实际制备过程中发现,产物中有以下杂质:
方法三,路线长,原料贵,witting反应收率仅有50%,水解反应收率20%,氧化反应60%,总收率6%,成本不具有优势,并且witting反应对产生了大量无价值的污染物,三废处理成本高。
技术实现要素:
本发明所要解决的技术问题在于:针对已报道的1,2,3,4-四氢-1-萘甲酸合成方法的工艺难点,本发明的目的在于提供一种步骤短,收率高,原料便宜并且可工业化的1,2,3,4-四氢-1-萘甲酸合成方法。
为解决上述技术问题,本发明的以超强碱法制备1,2,3,4-四氢-1-萘甲酸的方法,包括以下步骤:
(1)氮气保护下,使用四氢萘做原料,加入叔丁醇钾,正己烷做溶剂,滴加正丁基锂,制备拔氢产物:
(2)氮气保护下,将干燥的二氧化碳通入体系中,酸化,得到目标产物
在步骤(1)中,叔丁醇钾与正丁基锂的摩尔比为1:0.5~1.5,优选1:1.0。
在步骤(1)中,超强碱与四氢萘的摩尔比为1:0.5~2,优选1:1.2~1.5。
在步骤(1)中,反应温度为-70~30℃,优选-10~20℃。
在步骤(1)中,反应时间为0.5~4小时,选优2小时。
在步骤(2)中,反应温度为-80~0℃,优选-40~0℃。
在步骤(2)中,反应时间为1~10小时,优选1~3小时。
在步骤(2)中,萃灭酸选择硫酸,盐酸,优选硫酸。
以下为本发明的若干优选实施方式:
优选实施方式之一包括以下步骤:
(1)向容器中投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用;
(2)体系降温至-70℃,通入干燥的二氧化碳30分钟,体系放热,升温至-30℃,控温-30~-70℃,反应1小时,hplc跟踪,至原料残留10%以下;
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全;
再用50%液碱调ph大于11,分层,静置分层,正己烷层弃之,水层用200ml正己烷萃取3次,水层用10g活性炭脱色1小时,抽滤,用25%硫酸调ph小于1,冷却析出类白色晶体;烘干得成品。
优选实施方式之二包括以下步骤:
(1)50l反应釜中,装有温度计、氮气保护口器,投入3000g四氢萘,氮气保护,加入10kg正己烷,加入9.05kg叔丁醇钾,控温0℃,滴加2.5m正丁基锂10.9l,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用;
(2)100l反应釜中,装有温度计、氮气保护口器,氮气保护下,加入20kg干冰,将步骤(1)所得备用液用氮气压入干冰体系中,开始无须搅拌,等体系温度升到-50℃,开启搅拌,反应60分钟,hplc跟踪,至原料残留5%以下;
用10升水萃灭反应液,升至室温,搅拌30分钟,萃灭完全;
分层,静置分层,正己烷层弃之,水层用5kg正己烷萃取3次,水层用500g活性炭脱色1小时,抽滤,用25%硫酸调ph小于1,冷却析出类白色晶体;烘干得成品2.7kg。
优选实施方式之三包括以下步骤:
(1)1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用;
(2)体系降温至-70℃,滴加16.2g碳酸二甲酯,体系放热,升温至-30℃,控温-30~-70℃,反应30分钟,hplc跟踪,至原料残留20%以下;
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全;
正己烷层分别用100ml水洗,饱和氯化钠洗涤一次,干燥,浓干,蒸馏,得成品。
优选实施方式之四包括以下步骤:
(1)1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用;
(2)体系降温至-70℃,滴加16.5gdmf,体系放热,升温至-30℃,控温-30~-70℃,反应30分钟,hplc跟踪,至原料残留20%以下;
降温到-70℃,用25%稀硫酸萃灭反应液,至ph为5~6,升至室温,搅拌5分钟,萃灭完全;
正己烷层分别用100ml水洗,饱和氯化钠洗涤一次,浓干,加入200ml乙醇,滴加50g饱和亚硫酸氢钠溶液,室温搅拌过夜,抽滤,乙醇洗涤,得到1,2,3,4-四氢-1-萘甲醛的亚硫酸氢钠盐31g;
31g粗品加200ml甲叔醚,5%稀盐酸200ml,室温搅拌过夜,分层,100ml水,100ml饱和氯化钠洗涤,无水硫酸钠干燥浓干,得成品8.6g,收率35.8%。
本发明相比现有技术有以下优点:步骤短,原料便宜,收率高,可操作性高,重复性好,工业化方便。
附图说明
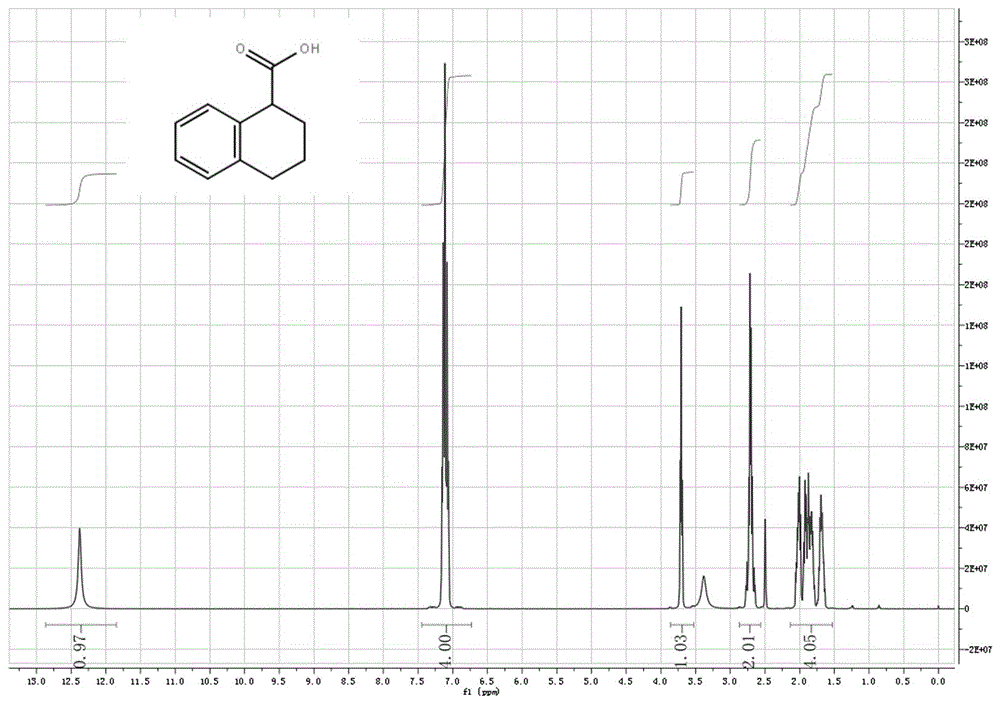
图1是实施例三所得产品的核磁谱图;
图2是实施例三所得产品的hplc谱图。
具体实施方式
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
所有化学品均为市售化学品。
1,2,3,4-四氢-1-萘甲酸的制备
实施例一:
1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
体系降温至-70℃,通入干燥的二氧化碳30分钟,体系放热,升温至-30℃,控温-30~-70℃,反应1个小时,hplc跟踪,至原料残留10%以下。
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全。
再用50%液碱调ph大于11,分层,静置分层,正己烷层弃之,水层用200ml正己烷萃取3次,水层用10g活性炭脱色1小时,抽滤,用25%硫酸调ph小于1,冷却析出类白色晶体。烘干得成品17.1g,收率:64.7%.
实施例二:
500ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
1000ml三口烧瓶中,装有温度计、氮气保护口器,氮气保护下,加入100g干冰,将备用液滴加体系中,搅拌反应30分钟,hplc跟踪,至原料残留5%以下。
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全。
再用50%液碱调ph大于11,分层,静置分层,正己烷层弃之,水层用200ml正己烷萃取3次,水层用10g活性炭脱色1小时,抽滤,用25%硫酸调ph小于1,冷却析出类白色晶体。烘干得成品19.1g,收率:72.3%.
实施例三:
50l反应釜中,装有温度计、氮气保护口器,投入3000g四氢萘,氮气保护,加入10kg正己烷,加入9.05kg叔丁醇钾,控温0℃,滴加2.5m正丁基锂10.9l,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
100l反应釜中,装有温度计、氮气保护口器,氮气保护下,加入20kg干冰,将备用液用氮气压入干冰体系中,开始无须搅拌,等体系温度升到-50℃,开启搅拌,反应60分钟,hplc跟踪,至原料残留5%以下。
用10升水萃灭反应液,升至室温,搅拌30分钟,萃灭完全。
分层,静置分层,正己烷层弃之,水层用5kg正己烷萃取3次,水层用500g活性炭脱色1小时,抽滤,用25%硫酸调ph小于1,冷却析出类白色晶体。烘干得成品2.7kg,收率:67.5%.
所得产品的核磁及hplc谱图请参见图1、图2。
同样的机理,我们还制备了以下衍生物:
实施例四:
1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
体系降温至-70℃,滴加16.2g碳酸二甲酯,体系放热,升温至-30℃,控温-30~-70℃,反应30分钟,hplc跟踪,至原料残留20%以下。
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全。
正己烷层分别用100ml水洗,饱和氯化钠洗涤一次,干燥,浓干,蒸馏,得产品11g,收率38.5%。hnmr(cdcl3):δ=7.17-7.08(4h,m),3.83(1h,t,j=5.7hz),3.70(3h,s),2.87-2.71(2h,m),2.17-2.09(1h,m),2.03-1.92(2h,m),1.80-1.72(1h,m)。
实施例五:
1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
体系降温至-70℃,滴加26.5g碳酸二乙酯,体系放热,升温至-30℃,控温-30~-70℃,反应30分钟,hplc跟踪,至原料残留10%以下。
降温到-70℃,用25%稀硫酸萃灭反应液,至ph小于1,升至室温,搅拌30分钟,萃灭完全。
正己烷层分别用100ml水洗,饱和氯化钠洗涤一次,干燥,浓干,蒸馏,得产品13g,收率42.4%。1h-nmr(cdcl3,400mhz)δh7.19-7.09(m,4h),4.18(q,2h,j7.2hz),3.81(t,1h,j6.0hz),2.88-2.72(m,2h),2.18-2.09(m,1h),2.05-1.94(m,2h),1.80-1.72(m,1h),1.27(t,3h,j7.2hz).
实施例六:1000ml三口烧瓶中,装有温度计、氮气保护口器,投入19.8g四氢萘,氮气保护,加入200ml正己烷,加入59.6g叔丁醇钾,控温0℃,滴加2.5m正丁基锂72ml,放热,控温10℃以下,滴毕,10~20℃反应1小时,备用。
体系降温至-70℃,滴加16.5gdmf,体系放热,升温至-30℃,控温-30~-70℃,反应30分钟,hplc跟踪,至原料残留20%以下。
降温到-70℃,用25%稀硫酸萃灭反应液,至ph为5~6,升至室温,搅拌5分钟,萃灭完全。
正己烷层分别用100ml水洗,饱和氯化钠洗涤一次,浓干,加入200ml乙醇,滴加50g饱和亚硫酸氢钠溶液,室温搅拌过夜,抽滤,乙醇洗涤,得到1,2,3,4-四氢-1-萘甲醛的亚硫酸氢钠盐31g(包含少量亚硫酸氢钠)。
31g粗品加200ml甲叔醚,5%稀盐酸200ml,室温搅拌过夜,分层,100ml水,100ml饱和氯化钠洗涤,无水硫酸钠干燥浓干,得成品8.6g,收率35.8%。1hnmr(400mhz,chloroform-d)δ9.68(d,j=2.2hz,1h),7.24–7.18(m,2h),7.19–7.12(m,2h),3.64–3.53(m,1h),2.79(t,j=6.3hz,2h),2.28–2.19(m,1h),2.00–1.85(m,1h),1.88–1.74(m,2h).
以上所述仅为本发明的较好实施例,并不用以限制本发明,凡在本发明的精神和原则之内所做的任何修改、等同替换和改进等,均应包含在本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!