碳酸酯连接的表面改性大分子的制作方法
相关申请
这是一篇专利合作条约申请,其要求基于2018年3月9日提交的美国临时专利申请62/640,839的优先权的35u.s.c.§119的权益,该美国临时专利申请以其整体并入本文中。
本发明涉及表面改性大分子(smm)及其与基础聚合物的掺混物。所述掺混物可用在其中需要增强的表面性能(例如,降低或防止生物结垢、生物分子的固定或某些生物分子的变性的表面性能)的应用中,例如在工业和医学应用中。
背景技术:
润湿的表面可能易于与诸如蛋白质、核酸和活生物体的生物试剂相互作用。这些相互作用可导致生物试剂(例如蛋白质或核酸)的吸附的降解。这些相互作用还可能导致由水成分造成的表面结垢,所述水成分例如生物分子、活生物体(例如细菌)、溶解的无机或有机化合物、胶体和悬浮固体。生物结垢可归因于累积的细胞外材料,例如可溶性微生物产物和细胞外聚合物物质,例如多糖和蛋白质(参见,例如,asatekin等人,journalofmembranescience,285:81-89,2006)。例如,用于工业水过滤或用于医学应用中(例如透析中)的膜可能会由于例如蛋白质的吸附、悬浮粒子或沉淀的盐附接到膜上而遭到结垢。生物医学应用中结垢的其它一些实例通常可能由例如细胞和病原体附着到医疗设备(例如导管或其它可植入医疗设备)的表面而引起,并且这种结垢可能具有潜在不利的结果。结垢在海船船体上也可能是明显的,所述海船船体可能被海洋生物体或它们的分泌物所覆盖。
因此,具有减少或防止生物结垢、生物分子的固定或某些生物分子的变性的表面性能的组合物和掺混物可用于工业和医学的各种应用中。
技术实现要素:
发明概述
本发明提供碳酸酯连接的表面改性大分子。
在一个方面,本发明提供式(i)的化合物:
ft–oc(o)o–b–oc(o)o–[a–oc(o)o–b]n–oc(o)o–ft(i)
其中
(i)a包含软链段并经由碳酸酯键共价键合于b;
(ii)b包含聚环氧烷或由下式描述的结构部分:
并经由碳酸酯键共价键合于a;和
(iii)ft是包含多氟有机基基团的表面活性基团,其中ft经由碳酸酯键共价键合于b;和
(iv)n是1至10的整数。
在式(i)的一些实施方案中,所述化合物具有式(ii)的结构:
ft–oc(o)o–(ch2ch2o)m–oc(o)o–[a–oc(o)o–(ch2ch2o)m]n–oc(o)o–ft(ii)
其中
(i)a包含软链段;
(ii)ft是包含多氟有机基基团的表面活性基团;
(iii)m是2至4的整数;和
(iv)n是1至10的整数。
在式(i)中,b可含有聚环氧丙烷、聚环氧乙烷或聚四氢呋喃。在式(i)中,b可由三乙二醇,四乙二醇或双酚a形成。
在式(i)或(ii)中,a可含有氢化聚丁二烯(hlbh)、氢化聚异戊二烯(hhtpi)、聚((2,2-二甲基)-1,3-亚丙基碳酸酯)、聚丁二烯、聚(二乙二醇)己二酸酯(pega)、聚(六亚甲基碳酸酯)(phcn)、聚(乙烯-共聚-丁烯)、(二乙二醇-邻苯二甲酸酐)聚酯、(1,6-己二醇-邻苯二甲酸酐)聚酯、(新戊二醇-邻苯二甲酸酐)聚酯(pdp)、聚硅氧烷、双酚a乙氧基化物、聚(环氧乙烷)-b-聚(环氧丙烷)-b-聚(环氧乙烷)(pln)、聚环氧乙烷(peo)、聚环氧丙烷(ppo)或聚四氢呋喃(ptmo)。
在式(i)或(ii)的一些实施方案中,a不包括酯键。例如,a包括氢化聚丁二烯(hlbh)、氢化聚异戊二烯(hhtpi)、聚((2,2-二甲基)-1,3亚丙基碳酸酯)、聚丁二烯,聚(乙烯-共聚-丁烯)、聚硅氧烷、双酚a乙氧基化物、聚(环氧乙烷)-b-聚(环氧丙烷)-b-聚(环氧乙烷)(pln)、聚环氧乙烷(peo)、聚环氧丙烷(ppo)或聚四氢呋喃(ptmo)。
在式(ii)中,a可含有三嵌段共聚物ppo-b-peo-b-(聚硅氧烷)-b-peo-b-ppo(plnsi)。在式(ii)中,a可含有氢化聚异戊二烯(hhtpi)或氢化聚丁二烯(hlbh)。在式(ii)中,a可含有聚环氧丙烷(ppo)或聚四氢呋喃(ptmo)。在式(ii)中,a可含有聚环氧乙烷-聚二甲基硅氧烷-聚环氧乙烷(c10mwpeo=2,500da),聚环氧乙烷-聚二甲基硅氧烷-聚环氧乙烷(c15mwpeo=1,000da)或聚环氧乙烷-聚二甲基硅氧烷-聚环氧乙烷(c22mwpeo=2,500da)。在式(ii)中,a可含有环氧丙烷-聚二甲基硅氧烷-环氧丙烷嵌段共聚物(c22mwppo=2,500da)。在式(ii)中,a可含有聚环氧乙烷(peo)。在式(ii)中,a可含有二乙二醇-邻苯二甲酸酐。在式(ii)中,a可含有聚(环氧乙烷)-b-聚(环氧丙烷)-b-聚(环氧乙烷)(pln)。
在另一方面,本发明提供式(iii)的化合物:
其中
(i)ft是多氟有机基基团;
(ii)x1和x2中的每个独立地是h、ch3或ch2ch3;
(iii)b包含聚环氧烷;和
(v)n是5至100的整数。
在一个相关方面,本发明提供式(iv)的化合物:
其中
(i)每个ft是多氟有机基;
(ii)x1和x2中的每个独立地是h、ch3或ch2ch3;
(iii)b包含聚环氧烷;和
(iv)n1和n2中的每个独立地是5至50的整数。
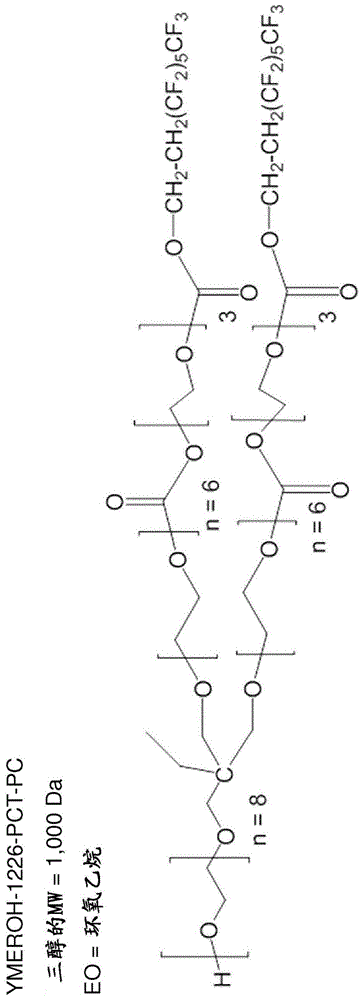
在式(iii)或(iv)中,b可含有聚环氧丙烷、聚环氧乙烷或聚四氢呋喃。在式(iii)或(iv)中,b可由三乙二醇或四乙二醇形成。在式(iii)中,b可以是聚环氧乙烷,x1是乙基,和x2是h(ymeroh-1226-pct-pc)。在式(iii)中,b可以是聚环氧乙烷,x1是乙基,和x2是甲基(ymer-1226-pct-pc)。在式(iv)中,b可以是聚环氧乙烷,x1是h,和x2是h(xmer-1226-pct-pc)。
在式(i)、(ii)、(iii)或(iv)中,ft可以是以下通式的基团:
chmf(3-m)(cf2)rch2ch2—和chmf(3-m)(cf2)s(ch2ch2o)x—,其中
m是0、1、2或3;
x是1-10之间的整数;
r是2-20之间的整数;和
s是1-20之间的整数。
在一些实施方案中,m是0或1。
在式(i)、(ii)、(iii)或(iv)中,所述化合物可具有小于10,000da的理论分子量。
定义
如本文中使用的,术语“烷基”是指具有1至10个碳原子(c1-10)的支化或非支化饱和烃基团。烷基可任选包括单环、双环或三环的环体系,其中每个环理想地具有三至六个成员。所述烷基基团可以是未取代的或被1、2或3个取代基取代的,所述取代基独立地选自烷氧基、芳氧基、烷硫基、芳硫基、卤素、二取代的氨基和酯。
如本文中使用的,术语“亚烷基”是指二价烷基基团。
如本文中使用的,术语“基础聚合物”是指具有大于或等于20kda(例如,大于或等于50kda,大于或等于75kda,大于或等于100kda,大于或等于150kda,或大于200kda)的理论分子量的聚合物。基础聚合物的非限制性实例包括聚氨酯、聚砜、聚碳酸酯、聚酯、聚酰胺、聚酰亚胺、聚亚烷基(alkylene)(例如,聚乙烯、聚丙烯、聚苯乙烯、聚丁二烯、聚异戊二烯、聚(丙烯腈-丁二烯-苯乙烯)、聚甲基丙烯酸甲酯、聚乙酸乙烯酯、聚丙烯腈、聚氯乙烯)、有机硅聚合物、多糖(例如纤维素、乙酸纤维素、二乙酸纤维素或三乙酸纤维素)和它们的共聚物(例如聚对苯二甲酸乙二醇酯)。
如本文中使用的,术语“碳酸酯键(carbonatelinkage)”是指碳酸的酯。
如本文中使用的,术语“分子量”是指阿伏加德罗(avogadro)数的相同组成的分子的理论重量。由于表面改性大分子的制备可能涉及产生一定分布的化合物,因此术语“分子量”是指由反应性成分的化学计量确定的理想化结构。因此,如本文中使用的,术语“分子量”是指理论分子量。
如本文中使用的,术语“低聚链段”是指小于约200个单体单元的一定长度的一种或多种重复单元。低聚链段可具有小于或等于10,000da,但优选<7,000da和在一些实例中<5,000da的理论分子量。本发明的表面改性大分子可以由低聚链段二醇、三醇或四醇形成,以得到式(i)、(ii)、(iii)或(iv)的化合物。低聚链段的非限制性实例包括聚环氧烷(例如聚环氧乙烷)、氢化聚丁二烯、氢化聚异戊二烯、聚((2,2-二甲基)-1,3-亚丙基碳酸酯)、聚丁二烯、聚(二乙二醇)己二酸酯、聚(六亚甲基碳酸酯)、聚(乙烯-共聚-丁烯)、(二乙二醇-邻苯二甲酸酐)聚酯、(1,6-己二醇-邻苯二甲酸酐)聚酯、(新戊二醇-邻苯二甲酸酐)聚酯、聚硅氧烷和双酚a乙氧基化物。
当在本文中关于基础聚合物使用时,术语聚亚烷基(polyalkylene)”是指一种基础聚合物,其由具有2至4个碳原子的直链或支化亚烷基重复单元和/或任选的3至10个碳原子的环状烯烃(例如降冰片烯)组成。每个亚烷基重复单元任选被一个选自氯、甲氧基羰基、乙氧基羰基、羟基、乙酰氧基、氰基和苯基的取代基取代。聚亚烷基基础聚合物可以是共聚物(例如,甲基丙烯酸甲酯-丙烯腈-丁二烯-苯乙烯(mabs)、甲基丙烯酸甲酯-丁二烯-苯乙烯(mmbs)、甲基丙烯酸酯-丁二烯-苯乙烯(mbs)、苯乙烯-丁二烯(sb)、苯乙烯-丙烯腈(san),苯乙烯-甲基丙烯酸甲酯(smma)、环状烯烃共聚物(coc)或环状烯烃聚合物(cop)共聚物)。聚亚烷基基础聚合物的非限制性实例包括聚苯乙烯、cop、coc、mabs、san、smma、mbs、sb和聚丙烯酸酯(例如,pmma)。
如本文中使用的,术语“聚醚砜”意思是下式的聚合物:
聚醚砜(pes)
该聚合物可以商品名radeltm商购自amococorp.。
如本文中使用的,术语“多氟有机基基团”是指一种烃基团,在该烃基团中2至59个氢原子被氟原子替代。所述多氟有机基基团含有1至30个碳原子。所述多氟有机基基团可含有直链烷基、支化烷基或芳基基团,或它们的任何组合。所述多氟有机基基团可以是“多氟酰基”,在该基团中,所述多氟有机基基团(例如,多氟烷基)附接至所述分子的其余部分所通过的碳原子被氧代取代。在多氟有机基基团(例如多氟烷基)内的烷基链可被最高至9个氧原子间断,条件是在多氟有机基内两个最接近的氧原子被至少2个碳原子隔开。当所述多氟有机基由任选被氧代取代的和/或任选被氧原子间断的直链或支化烷基组成时,如本文中定义的那样,这样的基团可被称为多氟烷基基团。一些多氟有机基基团(例如,多氟烷基)可具有100da至1,500da的理论分子量。多氟烷基可以是cf3(cf2)r(ch2ch2)p—,其中p为0或1,r为2至20,或者是cf3(cf2)s(ch2ch2o)x—,其中x为0至10,和s为1至20。或者,多氟烷基可以是chmf(3-m)(cf2)rch2ch2—或chmf(3-m)(cf2)s(ch2ch2o)x—,其中m为0、1、2或3;x为0至10;r为2至20的整数;和s为1至20的整数。在特定的实施方案中,x为0。在其它实施方案中,多氟烷基是全氟庚酰基。在某些实施方案中,多氟烷基是由以下物质形成的:1h,1h,2h,2h-全氟-1-癸醇;1h,1h,2h,2h-全氟-1-辛醇;1h,1h,5h-全氟-1-戊醇;或1h,1h-全氟-1-丁醇,和它们的混合物。在再其它一些实施方案中,多氟烷基是(cf3)(cf2)5ch2ch2o—、(cf3)(cf2)7ch2ch2o—、(cf3)(cf2)5ch2ch2o—、chf2(cf2)3ch2o—、(cf3)(cf2)2ch2o—或(cf3)(cf2)5—。在再其它一些实施方案中,所述多氟烷基基团含有(cf3)(cf2)5—。
所述“聚(氧基-1,4-亚苯基磺酰基-1,4-亚苯基氧基-1,4-亚苯基异丙叉基-1,4-亚苯基)”意思是下式的聚合物:
该聚合物可以商品名udeltmp-3500商购自solvayadvancedpolymers。
如本文中使用的术语“聚砜”是指一类聚合物,其包括作为重复子单元的结构部分“-芳基-so2-芳基-”。聚砜包括但不限于聚醚砜和聚(氧基-1,4-亚苯基磺酰基-1,4-亚苯基氧基-1,4-亚苯基异丙叉基-1,4-亚苯基)。
所述“表面活性基团”意思是与表面改性大分子共价链接的亲脂基团。可将表面活性基团定位以封住所述表面改性大分子的中心聚合物部分的一个、两个、三个或四个末端。表面活性基团包括多氟有机基基团(例如,多氟烷基和氟化聚醚)及其组合。
如本文中使用的术语“表面改性大分子”是指本文中描述的大分子(例如,根据式(i)-(iv)中任一种的化合物,例如,化合物(1)-(9)中任一种的化合物)。
如本文中使用的术语“热降解温度”是指在热重分析过程中开始发生表面改性大分子的至少5%(重量/重量)的重量损失的最低温度。
从附图、发明详述和权利要求书中,本发明的其它特征和优点将显而易见。
附图说明
图1显示了化合物(1)的结构。
图2显示了化合物(2)的结构。
图3显示了化合物(3)的结构。
图4显示了化合物(4)的结构。
图5显示了化合物(5)的结构。
图6显示了化合物(6)的结构。
图7显示了化合物(7)的结构。
图8显示了化合物(8)的结构。
图9显示了化合物(9)的结构。
发明详述
一般地,本发明提供表面改性大分子(smm),其结构基于通过具有至少一个碳酸酯键的连接基将低聚链段连接至表面活性基团。本发明的表面改性大分子可具有本文中描述的式(i)-(iv)中任一种的结构(例如,本发明的表面改性大分子可以是化合物(1)-(9)中的任一种)。
本发明提供碳酸酯连接基,其可在基础聚合物中引入亲水性表面能,尽管高度疏水的氟代烷基端基向表面迁移,但经历重排以暴露在所述碳酸酯连接基中存在的亲水性环氧乙烷基团。
典型地,氨基甲酸酯是氢供体,其由于自氢键化而倾向于聚集在一起。结果,这样的氨基甲酸酯可具有高的接触角,主要是疏水的,在与水相互作用方面提出挑战,这是使用smm向表面赋予抗生物结垢性能的重要标准。相反,本发明描述了亲水性化合物,其包括与碳酸酯键组合的聚环氧乙烷单元(例如,图1)。与传统聚氨酯相比,碳酸酯连接的smm的亲水特征可能是重要的,因为它们吸引水并提供抗生物结垢特性。
与其中碳酸酯键被酯键代替的相应化合物相比,本发明的化合物可以是水解稳定的。
特别地,本发明提供了基础聚合物与表面改性大分子的掺混物以及由其制备的制品。在一些实施方案中,在与基础聚合物形成的掺混物中使用多于一种的包括碳酸酯的smm。相对于缺乏表面改性大分子的制品,本发明的制品可表现出有利的表面性能。例如,可以使表面性能改性以使这种表面具有抗生物结垢性、生物分子的固定性或生物分子变性的介导作用。生物结垢可归因于累积的细胞外物质,例如可溶性微生物产物和细胞外聚合物物质,例如多糖和蛋白质(例如,asatekin等人,journalofmembranescience,285:81-89,2006)。特别地,本发明的表面可以抗结垢(例如,生物结垢)。本发明的表面还可以减少生物试剂(例如,多肽(例如,单克隆抗体或其抗原结合片段)、多核苷酸(例如,sirna或反义化合物)或疫苗)的降解(例如,通过吸附或变性);所述降解可能是由于在所述生物试剂与缺乏表面改性大分子的表面之间的相互作用导致的。不受理论的束缚,包括有所述表面改性大分子可以改变表面润湿性(与水),从而减少在生物试剂(例如蛋白质、核酸或细菌)和所述表面之间的接触。本发明的表面可能能够维持与生物制品的延长接触而不会引起实质的变性或固定,例如,所述生物制品可以是阿巴西普(abatacept),干扰素β-1a或胰岛素。特别地,这些和其它生物制品可得益于本发明的表面性能,该表面性能减少或防止了在所述表面和所述生物制品之间不希望的相互作用(例如,所述生物制品的固定和/或变性)。或者,包括有所述表面改性大分子可增加表面润湿性(与水)。这样的材料可用于需要亲水表面的应用中。
据信在本发明制品中的理想表面性能是由本发明的表面改性大分子提供的,本发明的表面改性大分子在制造过程中迁移到所述制品的表面,从而在所述制品的表面处暴露所述表面活性基团。所述表面活性基团可能部分负责将所述表面改性大分子携带到掺混物的表面,在那里,所述表面活性基团暴露在所述表面上。所述表面改性大分子向所述表面的迁移是一个动态过程,并取决于表面环境。迁移的过程受到在混合物表面处倾向于建立低表面能的趋势所驱动。当在锚固和表面迁移之间达到平衡时,所述表面改性大分子在聚合物表面处保持稳定,而同时改变了表面性能。可以通过低聚链段提供在基础聚合物内的锚定。
多个低聚物分子的聚集可增加它们的有效分子半径,从而降低了低聚物分子穿过基础聚合物的渗透性。通过本发明的表面改性大分子可改进表面性能改性的有效性。通过排除在相同分子内的氢键供体和受体的组合,由于聚集可能减少,本发明的表面改性大分子迁移到制品的表面的能力可获得增强。此外,与具有酯键的smm相比,包括碳酸酯键的smm可表现出增加的稳定性,这是由于对碳酸酯键的水解降解而言更大的稳定性所导致的。本发明的表面改性大分子可表现出增强的迁移到制品表面的能力,而不会损害它们在基础聚合物中的锚定。因此,本发明的某些表面改性大分子不含有氢键供体(例如,o-h、n-h或s-h结构部分)。特别地,所述表面改性大分子可能不含氨基甲酸酯结构部分。
对特定smm和特定基础聚合物的组合进行的选择可由许多因素来确定。首先,待添加到基础聚合物中的smm的类型和量部分地取决于所述掺混物是否形成单一稳定相,其中所述smm可溶于所述基础聚合物中(例如,分离所述掺混物以形成两个或更多个不同的相会指示不稳定的溶液)。然后,可以通过各种已知的分析方法测试所述掺混物的相容性。可以通过任何可用的光谱方法,例如具有元素分析(ea)的x射线光电子能谱(xps),来分析作为膜或作为纤维的掺混物的表面。得自xps的数据可指示出由迁移性smm改性所述表面的程度,和得自ea的数据可指示出本体材料的改性程度。然后可以测试稳定的掺混物,以确定在各种条件下表面的抗结垢性能。
在特定的实施方案中,所述表面改性可以保持相对于纯基础聚合物的透明性。包括有在基础聚合物中的掺混物经常可能导致损害的光学性能(例如较低的透明性),从而限制了这样的材料在其中需要材料透明性的应用中的利用。相反,包括表面改性大分子和基础聚合物的本发明制品可具有与纯基础聚合物相同或略低的透明性。
本发明的制品可至少部分地使用需要高温加工的工艺(例如,挤出或模制)由基础聚合物制备。例如,coc和cop经常需要大于200℃(例如,大于或等于250℃,或大于或等于300℃)的加工温度。本发明的一些化合物,例如pdp-1226pct(tg=314℃),适于高温加工。本文中描述的表面改性大分子可以是热稳定的(例如,可具有大于或等于200℃(例如,大于或等于250℃或大于或等于300℃)的热降解温度)。因此,本发明的制品可由基础聚合物和表面改性大分子的掺混物在大于200℃(例如,大于或等于250℃或大于或等于300℃)的温度下形成。本发明的制品可由基础聚合物和表面改性大分子的掺混物制造(例如,通过高温加工,例如熔融加工)。可以在所述基础聚合物的熔融加工之前添加所述表面改性大分子以制备本发明的制品。为了通过熔融加工形成掺混物,可例如将所述表面改性大分子与粒化或粉末化的聚合物混合,和然后通过已知方法(例如模塑或熔融挤出)进行熔融加工。可将所述表面改性大分子直接与所述聚合物在熔融条件下混合,或者可以首先在布拉本德(brabender)混合器中以表面改性大分子/聚合物掺混物的母料形式与所述聚合物预混合。如果需要,可将所述表面改性大分子的有机溶液与粉末化或粒化的基础聚合物混合,随后蒸发溶剂,和然后进行熔融加工。或者,可在挤出成所需形状之前即刻将所述表面改性大分子注射到熔融的聚合物料流中以形成掺混物。
在熔融加工之后,可以进行退火步骤以在所述基础聚合物中增强本文中描述的有利性能的发展。附加于或代替退火步骤,还可以在两个加热辊之间将熔融加工的组合压花,两个加热辊中的一个或两个可以是图案化的。退火步骤通常在所述聚合物的熔融温度以下(例如,在约50℃至约220℃下)进行。
将所述表面改性大分子以足以实现特定应用所需的表面性能的量添加到基础聚合物中。通常,所用的表面改性大分子的量在所述掺混物的0.05-15%(重量/重量)范围内。所述量可以凭经验确定,并且可以根据需要或期望进行调整,以实现期望的表面性能而不会损害所述基础聚合物的其它物理性能。
表面改性大分子
本发明的表面改性大分子可以是式(i)、(ii)、(iii)和(iv)中的任一种的化合物。
本发明的表面改性大分子可以是式(i)的化合物:
ft–oc(o)o–b–oc(o)o–[a–oc(o)o–b]n–oc(o)o–ft(i),
其中
a包含软链段并经由碳酸酯键共价键合于b;
b包含聚环氧烷或由下式描述的结构部分:
并经由碳酸酯键共价键合于a;和
ft是包含多氟有机基基团的表面活性基团,其中ft经由碳酸酯键共价键合于b;和
n是1至10的整数。
特别地,式(i)的化合物可以是式(ii)的化合物:
ft–oc(o)o–(ch2ch2o)m–oc(o)o–[a–oc(o)o–(ch2ch2o)m]n–oc(o)o–ft(ii),
其中
a包含软链段;
ft是包含多氟有机基基团的表面活性基团;
m是2至4的整数;和
n是1至10的整数。
本发明的表面改性大分子可以是式(iii)的化合物:
其中
ft是多氟有机基基团;
x1和x2中的每个独立地是h、ch3或ch2ch3;
b包含聚环氧烷;和
n是5至100的整数。
本发明的表面改性大分子可以是式(iv)的化合物:
其中
每个ft是多氟有机基;
x1和x2中的每个独立地是h、ch3或ch2ch3;
b包含聚环氧烷;和
n1和n2中的每个独立地是5至50的整数。
低聚链段
本发明的表面改性大分子可以由低聚链段二醇、三醇或四醇制备。因为所述反应对水分敏感,所以它们通常是在惰性n2气氛下和在无水条件下进行的。可以在合适的情况下分离和纯化所得的表面改性大分子。式(iii)或(iv)的表面改性大分子可例如由可商购的单二羟基取代的烷基或烷氧基烷基封端的peg(例如,ymertmn120,一种双官能聚乙二醇单甲基醚,得自perstorp)制备。示例性的低聚链段二醇、三醇和四醇如下所示。
方案1显示了可用于制备式(iii)的表面改性大分子的低聚链段三醇的结构的非限制性实例:
方案1
polyol3165(perstorp)
三羟甲基丙烷乙氧基化物
mw=1,000da
方案2显示了一些可用于制备式(i)或(ii)的化合物的低聚链段二醇:
方案2
pln=普朗尼克(pluronics)
pdp二醇
方案3显示了一些可用于制备式(ii)化合物的低聚链段二醇:
方案3
c15硅氧烷二醇
mw=1,000da
羟基封端的聚二甲基硅氧烷(eto-pdms-oet)嵌段共聚物(m=1,n为整数)
c22硅氧烷二醇
mw=2,500-3,000da
羟基封端的聚二甲基硅氧烷(pro-pdms-opr)嵌段共聚物(m=
12-16,n为整数)
c25硅氧烷二醇
mw=3,500da
羟基封端的聚二甲基硅氧烷(eto-pdms-oet)嵌段共聚物(m=25,n为整数)
本领域中已知的二醇可用于制备式(i)或(ii)的化合物。例如,低聚链段的二醇可选自聚脲、聚氨酯、聚酰胺、聚环氧烷、聚碳酸酯、聚酯、聚内酯、有机硅聚合物、聚醚砜、聚亚烷基、聚乙烯基化合物、多肽多糖或它们的醚连接的或胺连接的链段(例如,所述链段在这种情况下可以指所列出的低聚物中的重复单元)。
合成
本发明的化合物可使用与在实施例中描述的那些方法类似的方法从适当选择的试剂(例如二羧酸衍生物、二醇和氟化醇)开始进行制备,以形成宽范围的基于碳酸酯的表面改性大分子。
制品
本发明还提供由本发明的掺混物形成的制品。可使用本发明的掺混物形成的制品包括但不限于手术帽、手术床单、手术盖布、手术罩衣、口罩、手套、手术帘、过滤器(例如呼吸器的部分、水过滤器、空气过滤器或面罩)、电缆、薄膜、面板、管道、纤维、片材和可植入医学装置(例如,心脏辅助装置、导管、支架、假体植入物、人工括约肌或药物递送装置)。
本发明的表面改性剂和掺混物可用于薄膜和非织造应用中,例如,手术帘、罩衣、面罩、包裹物、绷带和用于医学技术人员的其它防护性穿戴服装(例如,工作服、实验室外套)需要经常超过200℃的高温加工,形式为挤出制品(例如热塑性薄膜、热塑性纤维、纤维质非织造材料、热塑性泡沫材料等),其中加工温度可达到250-300℃的范围。在特定的实施方案中,在非织造应用中使用的表面改性剂是由双酚a形成的。本发明的表面改性剂和掺混物还可用于可植入的医学装置(例如,中央静脉导管以赋予降低的闭塞性能和增强的血液相容性)。本发明的表面改性剂和掺混物还可在用于流体或气体分离的由聚乙烯、聚丙烯或聚硅氧烷基础聚合物制成的中空纤维膜过滤中使用。
本发明的表面改性剂和掺混物可以在非织造织物制造中在加工过程中具有所需的高温稳定性,或者与在导管制造中使用的聚合物具有相容性。本发明的掺混物在加工过程中可具有所需的高温稳定性。在特定的实施方案中,适合于高温加工的表面改性剂是由双酚a形成的。因此,所述掺混物可提供所需的在高温下抗降解性,同时提供希望的生物相容性能,例如抗生物结垢性、抗生物分子固定在表面上的抗性和对介导生物分子变性的抗性。该技术可包括将smm并入到基础聚合物中,其然后再析出(bloom)至表面,因此改性了所述聚合物的表面,但保持本体性能完好。现在所述基础聚合物的氟化表面具有高度的疏水性。可由本发明的掺混物形成的制品包括可以是透皮或经皮的植入的医学装置。
植入的装置
可由本发明的掺混物形成的装置包括植入的装置。植入的装置包括但不限于假体,例如起搏器,电导线,例如起搏导线,除颤器,人造心脏,心室辅助装置,解剖重建假体,例如乳房植入物,人造心脏瓣膜,心脏瓣膜支架,心包补片,手术补片,冠状动脉支架,血管移植物,血管和结构性支架,血管或心血管分流器,生物导管,纱布(pledge),缝合线,瓣环成形术环,支架,钉(staple),瓣膜移植物,伤口愈合用真皮移植物,骨科脊柱植入物,骨科用针,子宫内装置,泌尿系统支架,最大面部重建板,牙齿植入物,人工晶状体,夹子,胸骨线,骨骼,皮肤,韧带,肌腱,和它们的组合。透皮装置包括但不限于各种类型的导管,插管,引流管,例如胸管,手术器械,例如钳子,牵开器,针和手套,以及导管套。皮肤装置包括但不限于烧伤敷料,伤口敷料和牙科硬件,例如牙桥支架和支撑组件。
如本文中描述的,用smm改性的医疗装置的示例性用途包括作为生物传感器,导管,心脏瓣膜,整形外科植入物,输尿管支架,通气管和药物递送装置的用途。在一个特定的实施方案中,在导管的制造中使用包括表面改性剂的掺混物,所述表面改性剂包括作为软链段的聚硅氧烷。
具体实施方式
实施例
缩写
ymer(二醇)=羟基封端的聚乙二醇单甲基醚
ymeroh(三醇)=三羟甲基丙烷乙氧基化物
ymertm=聚乙二醇单甲基醚二醇
xmer(四醇)=季戊四醇乙氧基化物
c10(二醇)=羟基封端的聚二甲基硅氧烷(环氧乙烷-pdms-环氧乙烷)嵌段共聚物(c10mwpeo=2,500da)
c25(二醇)=羟基封端的聚二甲基硅氧烷(环氧乙烷-pdms-环氧乙烷)嵌段共聚物(c25mwpeo=3,500da)
pln8k(二醇)=普朗尼克(pluronic)型(聚环氧乙烷-嵌段-聚环氧丙烷-嵌段-聚环氧乙烷),peo:ppo=80:20
pln(二醇)=普朗尼克型(聚环氧乙烷-嵌段-聚环氧丙烷-嵌段-聚环氧乙烷),peo:ppo=50:50
6plnsi(二醇)=羟基封端的普朗尼克型聚二甲基硅氧烷(ppo-peo-si-peo-ppo)嵌段共聚物,peo:ppo=75:25
16plnsi(二醇)=羟基封端的普朗尼克型聚二甲基硅氧烷(ppo-peo-si-peo-ppo)嵌段共聚物,peo:ppo=50:50
pdp=直链二乙二醇-邻苯二甲酸酐二醇
pega=聚(二(乙二醇己二酸酯))二醇
mabs=甲基丙烯酸甲酯-丙烯腈-丁二烯-苯乙烯
mmbs=甲基丙烯酸甲酯-丁二烯-苯乙烯
mbs=甲基丙烯酸酯-丁二烯-苯乙烯
sb=苯乙烯-丁二烯
san=苯乙烯-丙烯腈
smma=苯乙烯-甲基丙烯酸甲酯
表面改性大分子的制备
基于碳酸酯的表面改性大分子的一般合成描述
本发明的化合物可例如通过以下过程合成:使teg双氯甲酸酯与capstone62-al氟代醇反应以得到部分氟化的teg双氯甲酸酯-capstone62-al中间体,然后使其与软链段二醇、三醇或四醇反应以得到所需产物。另外,本发明的化合物可例如通过以下过程合成:使软链段二醇与双酚a氯甲酸酯反应以得到二醇-双酚a中间体,然后使其与capstone62-al氟代醇反应以得到期望产物。下面提供进一步的细节。
化合物1的合成
用于合成的所有玻璃器皿均在110℃下在烘箱中干燥过夜。
将20g(0.020mol)ymeroh三醇(mw=1000da)添加到200ml二颈烧瓶中,所述二颈烧瓶被脱气过夜,然后用n2吹扫。向配备有搅拌棒的500ml经烘箱干燥的二颈烧瓶中添加21.70g(0.079mol)三乙二醇(teg)双氯甲酸酯。将所述烧瓶脱气15分钟,和然后用干燥n2吹扫。在吹扫后,将62ml无水甲苯借助于插管转移到所述烧瓶中。搅拌所述teg双氯甲酸酯以溶解在溶剂中。现在将其在冰浴下冷却15分钟。向50ml加料漏斗中添加28.73g(0.079mol)capstone62-al氟代醇(1h,1h,2h,2h-全氟-1-辛醇),然后脱气15分钟并用干燥n2吹扫。向该加料漏斗中添加21ml无水甲苯,然后添加7g的无水吡啶,并摇动该漏斗以溶解所有试剂。将所述加料漏斗连接到所述反应烧瓶,并开始将capstone62-al滴加至经冷却的teg双氯甲酸酯的溶液中。在添加过程中,将搅拌保持在最低程度。在添加完成后,在室温(25℃)下在n2气氛下,使反应再进行10分钟,以形成部分氟化的teg双氯甲酸酯-capstone62-al中间体。在进行部分氟化的同时,将无水甲苯(125ml)经由插管添加到装有所述ymeroh三醇的烧瓶中,随后添加6g无水吡啶,并搅拌该混合物以溶解所述ymeroh三醇。将250ml加料漏斗连接到装有已部分氟化的teg双氯甲酸酯-capstone62-al中间体的500ml二颈烧瓶,并将所述ymeroh-三醇溶液经由插管转移到所述漏斗中。将所述ymeroh-三醇溶液以缓慢的连续料流添加到500ml容器中,直到所有ymeroh-三醇都被添加。将所述混合物在n2下在50℃下搅拌48小时。该反应产生大量白色吡啶盐,其在反应期间沉淀。所有添加和转移均在干燥n2气氛下进行以避免任何与空气的接触。
纯化包括使用whatman4滤纸真空过滤所述吡啶盐,随后旋转蒸发甲苯。用1nhcl处理产物,并将其在二氯甲烷-水混合物中萃取以除去过量的吡啶,然后用1nnaoh溶液中和。收集底部有机层,用蒸馏水洗涤两次,并然后将其旋转蒸发。在温和摇动下将粗产物(粘稠油)在250ml圆底烧瓶中与100ml蒸馏水在37℃下温育48小时,以除去未反应的ymeroh-三醇。倾析出含水的乳状上层,并将底部油在20%乙酸乙酯/己烷混合物中纯化三次。所述过程包括将油溶解在乙酸乙酯中,然后在冷己烷中沉淀。该过程除去了较低分子量的副产物,它们是teg-双氯甲酸酯和氟代醇反应的二和单氟化衍生物。
将经纯化的产物在75℃和4毫巴下干燥以形成粘稠的澄清油(42%收率)。通过gpc(基于聚苯乙烯标准物的分子量)和针对氟的元素分析表征所述经纯化的产物。平均分子量(聚苯乙烯当量)为4018g/mol。多分散度=1.24。元素分析显示%f=15%。首次开始时的热分解温度(tga,在n2下):223℃(在5重量%损失下)。化合物1(ymeroh-1226-pct-pc)的化学结构在图1中示出。
化合物2的合成
用于合成的所有玻璃器皿均在110℃下在烘箱中干燥过夜。
向配备有搅拌棒的200ml经烘箱干燥的二颈烧瓶中添加50g(0.048mol)ymer二醇(mw=1000da),并在60℃下在温和搅拌下脱气过夜。然后,将所述ymer二醇用干燥n2吹扫,然后使用插管将53ml无水氯仿(chcl3)添加到所述烧瓶中,随后添加15g无水吡啶。搅拌所述反应混合物以溶解所述试剂并获得均匀溶液。向配备有搅拌棒的1l经烘箱干燥的二颈烧瓶中添加60.9g(0.221mol)三乙二醇(teg)双氯甲酸酯。将所述烧瓶脱气15分钟,和然后用干燥n2吹扫。在吹扫后,将157ml无水chcl3借助于插管转移到所述烧瓶中。搅拌所述teg双氯甲酸酯以溶解在溶剂中。向500ml二颈烧瓶中添加73.59g(0.202mol)capstone62-al氟代醇(1h,1h,2h,2h-全氟-1-辛醇),然后脱气15分钟并用干燥n2吹扫。向其中添加314ml无水chcl3,随后添加28g吡啶。搅拌所述烧瓶以溶解所有试剂。使用插管将capstone62-al氟代醇溶液转移到500ml加料漏斗中,该漏斗事先已被脱气并用n2进行吹扫。将加料漏斗连接到在冰浴中冷却的装有所述teg双氯甲酸酯溶液的1l反应容器。在n2下逐滴添加所述氟代醇1小时。在反应期间将搅拌保持在最低程度,以形成部分氟化的teg双氯甲酸酯-capstone62-al氟代醇中间体。接下来,将所述ymer二醇溶液使用20口径的插管以缓慢连续的料流转移至1l反应容器中,同时将所述反应容器在冰浴下冷却直至所有ymer二醇溶液都已被添加。移去所述冰浴,并使反应在室温下再进行额外10分钟。然后将温度升至50℃,并使反应进行过夜。所有添加和转移均在干燥n2气氛下进行以避免任何与空气的接触。
通过以下过程纯化粗产物:首先在旋转蒸发仪上除去chcl3溶剂,将粗产物溶解在最少的thf中,和用冰浴冷却20分钟以沉淀吡啶盐。将所述溶液真空过滤,并使用旋转蒸发仪蒸发thf。用1nhcl处理产物,并将其在二氯甲烷-水混合物中萃取以除去过量的吡啶,然后用1nnaoh溶液中和。收集底部有机层,用蒸馏水洗涤两次,并然后将其旋转蒸发。最后,将产物溶于最小量的异丙醇(ipa)中,在己烷中沉淀出来,用己烷洗涤两次,并在真空下干燥。将产物在真空烘箱中在60℃下干燥过夜,以产生作为粘稠液体的产物(59%产率)。通过gpc(基于聚苯乙烯标准物的分子量)和针对氟的元素分析表征所述经纯化产物。平均分子量(聚苯乙烯当量)为3422g/mol。多分散度=1.15。元素分析:f=19%)。首次开始时的热分解温度(tga,在n2下):203℃(在5重量%损失下)。化合物2(ymer-1226-pct-pc)的化学结构在图2中示出。
化合物3的合成
用于合成的所有玻璃器皿均在110℃下在烘箱中干燥过夜。
向配有搅拌棒的200ml二颈烧瓶中添加12g(0.016mol)xmer四醇(mw=771da),在60℃下在温和搅拌下脱气过夜,并然后用n2吹扫。向配备有搅拌棒的500ml经烘箱干燥的二颈烧瓶中添加9g(0.033mol)三乙二醇(teg)双氯甲酸酯。将所述烧瓶脱气15分钟,和然后用干燥n2吹扫。在吹扫后,将65ml无水chcl3借助于注射器转移到所述烧瓶中。搅拌所述teg双氯甲酸酯以溶解在溶剂中。向50ml加料漏斗中添加15g(0.033mol)capstone62-al氟代醇(1h,1h,2h,2h-全氟-1-辛醇),并脱气15分钟和用干燥n2吹扫。向该加料漏斗中添加20ml无水chcl3,随后添加3g的无水吡啶,并且摇动所述烧瓶以溶解所有试剂。将所述加料漏斗连接到在冰中冷却的500ml反应烧瓶,并将capstone62-al氟代醇滴加到所述teg双氯甲酸酯中。所述添加的完成花费1小时,并使反应在室温(25℃)下在n2气氛下再进行额外10分钟,以形成部分氟化的teg双氯甲酸酯-capstone62-al中间体。在进行部分氟化的同时,将无水chcl3(122ml)经由插管添加到装有所述xmer四醇的烧瓶中,随后添加2.5g无水吡啶,并搅拌该混合物以溶解所述试剂。接下来,将所述x-mer四醇溶液使用20口径的插管以缓慢连续的料流转移至500ml反应容器中,同时将该反应容器在冰浴中冷却直至所有xmer二醇溶液都已被添加。移去所述冰浴,并使反应在室温下再进行额外10分钟。然后将温度升至50℃,并使反应进行过夜。所有添加和转移均在干燥n2气氛下进行以避免任何与空气的接触。
所述纯化包括从反应混合物中旋转蒸发chcl3、添加thf和通过真空过滤分离吡啶盐。用1nhcl处理产物,并将其在二氯甲烷-水混合物中萃取以除去过量的吡啶,然后用1nnaoh溶液中和。收集底部有机层,进一步用蒸馏水洗涤两次,并然后将其旋转蒸发。最后,将产物溶于最小量的异丙醇(ipa)中,在己烷中沉淀出来,用己烷洗涤两次,并在真空下干燥。将产物在真空烘箱中在60℃下干燥过夜,以产生作为粘稠液体的产物(59%产率)。
通过gpc(基于聚苯乙烯标准物的分子量)和针对氟的元素分析表征所述经纯化产物。平均分子量(聚苯乙烯当量)为2322g/mol。多分散度=1.12。元素分析显示f=25.8%。首次开始时的热分解温度(tga,在n2下):221℃(在10重量%损失下)。化合物3(xmer-1226-pct-pc)的化学结构在图3中示出。
化合物4的合成
用于合成的所有玻璃器皿均在110℃下在烘箱中干燥过夜。
向配备有搅拌棒的100ml经烘箱干燥的二颈烧瓶中添加15g(0.008mol)pdp二醇(mw=2000da),并在60℃下在温和搅拌下脱气过夜。然后,将所述pdp二醇用干燥n2吹扫,然后使用插管将90ml无水chcl3转移到所述烧瓶中,随后添加2g无水吡啶。搅拌所述反应混合物以溶解并获得均匀溶液。向50ml经烘箱干燥的加料漏斗中添加5.63g(0.016mol)双酚a氯甲酸酯,将漏斗脱气15分钟,并然后用干燥n2吹扫。在吹扫后,将45ml无水chcl3借助于插管转移到所述漏斗中。摇动所述漏斗以溶解所述氯甲酸酯。将所述加料漏斗连接到装有pdp二醇的烧瓶,并在室温下于1小时内滴加双酚a氯甲酸酯。使反应进行3小时,使得形成pdp-双酚a预聚物中间体。在进行预聚物中间体反应的同时,将7g(0.019mol)capstone62-al氟代醇添加到50ml二颈烧瓶中。将所述烧瓶脱气15分钟,和然后在干燥n2下吹扫。在用n2吹扫后,将15ml无水chcl3添加到所述烧瓶中,随后添加2g吡啶。摇动所述烧瓶以溶解所述氟代醇,然后将其使用插管以缓慢连续料流添加到装有所述预聚物中间体的200ml烧瓶中。将温度上升至60℃,并使最终的封端反应进行过夜。将产物通过以下过程纯化:将所述chcl3反应混合物倾倒到装有去离子水的分液漏斗中,并用5nhcl酸化含水层以中和任何残留的吡啶。
将产物萃取到用1nnaoh溶液中和的有机层中,并用去离子水洗涤两次。将有机层在无水na2so4上干燥。在旋转蒸发仪上除去溶剂,将固体残余物溶解在thf中,并将其在3:1水/甲醇混合物中沉淀。将产物在真空烘箱(30毫巴)中干燥2天以产生固体。通过gpc(基于聚苯乙烯标准物的分子量)表征所述经纯化产物。平均分子量(聚苯乙烯当量)为20704g/mol。多分散度=1.52。元素分析显示4.20重量%的f,首次开始时的热分解温度(tga,在n2下):314℃(在5重量%损失下)。化合物4(pdp-1226-pct-pc)的化学结构在图4中示出。
生物制品在表面上的固定和/或变性的测量
本发明制品的表面降低或预防生物制品的固定的能力可与由相同基础聚合物制备的但缺少表面改性大分子的制品的表面的所述能力相比较。在非限制性实例中,可以向由基础聚合物和表面改性大分子的掺混物制备的容器(“容器”)装入预定浓度的生物制品(例如干扰素β,单克隆抗体,融合蛋白(例如阿巴西普),sirna或dna(例如质粒))的溶液(例如水溶液)。然后可以例如在惰性气氛下(例如在ar或n2下)密封容器。在将生物制品溶液在密封容器内在室温下或在更低温度下(例如,在4℃或0℃下)在环境光(例如荧光灯)下或在黑暗中贮存一段时间(例如1天,3天,1周,2周,3周,1个月,2个月,3个月,0.5年,0.75年,1年等)后,可以评估贮存在容器内的溶液的总蛋白质或核酸浓度(例如,使用本领域已知的紫外-可见光谱仪或粒子分析仪)。然后可以将所述生物制品在容器内的浓度随时间的变化与所述生物制品在缺少表面改性大分子的容器(“对照容器”)内的浓度随时间的变化进行比较。经过相同的时间段,在容器中的所述生物制品浓度的降低幅度可以比在对照容器中的所述生物制品浓度的降低幅度低至少5%(例如,低至少10%,低至少20%,低至少30%,低至少40%或低至少50%),条件是所述溶液被贮存在相同的温度和光照条件下。
其它实施方案
所描述的本发明的材料和使用方法的各种改进和变化对本领域技术人员将是显而易见的,而不背离本发明的范围和主旨。尽管已经结合具体实施方案描述了本发明,但是应当理解,不应当将所要求保护的本发明不适当地限到这些具体实施方案上。实际上,对于本领域技术人员显而易见的所描述的用于实施本发明的方式的各种改进都意于在本发明的范围内。
其它实施方案在权利要求中。
- 还没有人留言评论。精彩留言会获得点赞!