氧化锌纳米晶、其制备方法、氧化锌纳米晶墨水和电致发光器件与流程
本发明涉及量子点材料领域,具体而言,涉及一种氧化锌纳米晶、其制备方法、氧化锌纳米晶墨水和电致发光器件。
背景技术:
随着科技的不断进步,量子点发光二极管(QLED)以其独特的优势逐渐兴起,并逐渐成为取代OLED(有机发光二极管)的新一代产品。量子点发光二极管(QLED)的电子传输层可以采用氧化锌来制备,但该项技术一直不够成熟,直到2010钱磊在Nano Today((2010)5,384~389)发表文章,将基于溶液法低温(30℃)合成的氧化锌(ZnO)纳米晶用于量子点发光二极管(QLED)的电子传输层,该方法主要利用二水合醋酸锌(Zn(Ac)2·2H2O)和四甲基氢氧化铵五水化合物(TMAH)作为合成原料,其得到的表面配体主要是醋酸根和羟基的氧化锌纳米晶,该发光二极管的外量子效率(EQE)可以达到近2%,开启电压为1.8伏特及1.7伏特,由于其具有比较好的导电性的优点,被后面的人们一直沿用。
但是实际上,现有的溶液法合成的氧化锌纳米晶虽然导电性好、无机材料对水敏感性低、稳定性高于有机传输层,但它的缺点也逐渐显现,尤其是由于缺陷态严重,ZnO层会出现缺陷复合放光的现象,这从荧光光谱(PL)中可以看出其缺陷发光已经占主导作用,并且该方法制备的发光二极管器件容易出现电子注入与空穴注入不平衡的现象,致使注入过快的电子被浪费,从而降低了器件的效率,这些都在一定程度上阻碍了量子点发光二极管推向实践应用的脚步。
技术实现要素:
本发明的主要目的在于提供一种氧化锌纳米晶、其制备方法、氧化锌纳米晶墨水和电致发光器件,以解决现有技术中氧化锌纳米晶的表面缺陷的问题。
为了实现上述目的,根据本发明的一个方面,提供了一种氧化锌纳米晶,该氧化锌纳米晶具有表面配体,配体为其中,R1、R2和R3各自独立地选自C1~10的烷基中的任意一种。
进一步地,上述配体为三乙基硫膦、三丁基硫膦和三辛基硫膦中的任意一种或多种,优选氧化锌纳米晶的表面配体内晶体的粒径为5~20nm,更优选为5~10nm。
根据本申请的另一方面,提供了一种上述氧化锌纳米晶的制备方法,该制备方法包括:步骤S1,采用溶液法制备初始氧化锌纳米晶的溶液,初始氧化锌纳米晶具有羧酸根表面配体;步骤S2,使溶液与硫前驱体在50~200℃下反应,生成氧化锌纳米晶,硫前驱体为S的溶液,其中,R1、R2和R3各自独立地选自C1~10的烷基中的任意一种,且在反应过程中控制反应体系的pH值在7~10之间。
进一步地,上述硫前驱体为S的溶液,其中,R1、R2和R3为C1~4的烷基中的任意一种,步骤S2包括:使溶液与硫前驱体在50~160℃下反应10~60min,生成氧化锌纳米晶。
进一步地,上述硫前驱体为S的溶液,其中,R1、R2和R3为C5~10的烷基中的任意一种,步骤S2包括:使溶液与硫前驱体在160~200℃下反应10~60min,生成氧化锌纳米晶。
进一步地,上述步骤S1中,硫前驱体中的硫与溶液中初始氧化锌纳米晶的摩尔比为10:1~100:1。
进一步地,上述步骤S1中,溶液为醇溶液或醚溶液,优选醇溶液为乙醇溶液、乙二醇溶液或丙二醇溶液,优选醚溶液为丙二醇甲醚溶液。
进一步地,上述步骤S2包括:步骤S21,使初始氧化锌纳米晶的溶液与硫前驱体在50~200℃下反应,得到含有氧化锌纳米晶的产物体系;步骤S22,提纯产物体系中的氧化锌纳米晶。
进一步地,上述步骤S22包括:步骤S221,将产物体系与乙酸乙酯混合后进行第一次固液分离,得到下层沉淀物,即得到氧化锌纳米晶,优选采用离心进行第一次固液分离;及可选的以下步骤,步骤S222,将沉淀物与乙醇、乙醇胺混合后向其中加入乙酸乙酯至混合体系变白色,得到待分离液;步骤S223,对待分离液进行第二次固液分离,得到位于下层的具有乙醇胺配体修饰的氧化锌纳米晶,优选采用离心进行第二次固液分离。
进一步地,上述步骤S22包括:加入醇和/或酮至产物体系中进行洗涤,得到含氧化锌纳米晶的洗涤液,其中,优选醇选自乙醇、甲醇、乙二醇和丙二醇中的任意一种或多种,酮为丙酮;加入乙酸乙酯至洗涤液中,分离出沉淀,得到纯化的氧化锌纳米晶。
根据本发明的另一方面,提供了一种氧化锌纳米晶墨水,包括氧化锌纳米晶和溶剂,该氧化锌纳米晶为上述的氧化锌纳米晶。
进一步地,上述溶剂为乙醇、丁醇、庚醇、癸醇、乙二醇、丙二醇、丙二醇醚类、丙二醇酯类中任一种或几种。
根据本发明的又一方面,提供了一种电致发光器件,包括电子传输层或/和电子注入层,该电子传输层或/和电子注入层采用上述的氧化锌纳米晶墨水制备而成。
应用本发明的技术方案,氧化锌纳米晶的表面配体和氧化锌纳米晶形成一层包覆的ZnS,既消弱了氧化锌纳米晶体的主要来自其表面Zn的悬挂键导致的缺陷发光,又可以通过调节硫的前驱体种类来方便的调节氧化锌纳米晶的表面配体,进而调节ZnO的电子迁移率。以三丁基硫膦为例,由于氧化锌纳米晶表面的羧酸根表面配体换成了空间位阻更大的三丁基硫膦,增大了相邻氧化锌纳米晶的距离,从而减小了氧化锌纳米晶的电子迁移率,在一定程度上缓解了电子和空穴注入的不平衡问题,本申请的氧化锌纳米晶所形成的氧化锌纳米晶体膜的电学性质得到了改进,进而解决了量子点发光二极管的缺陷发光问题。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
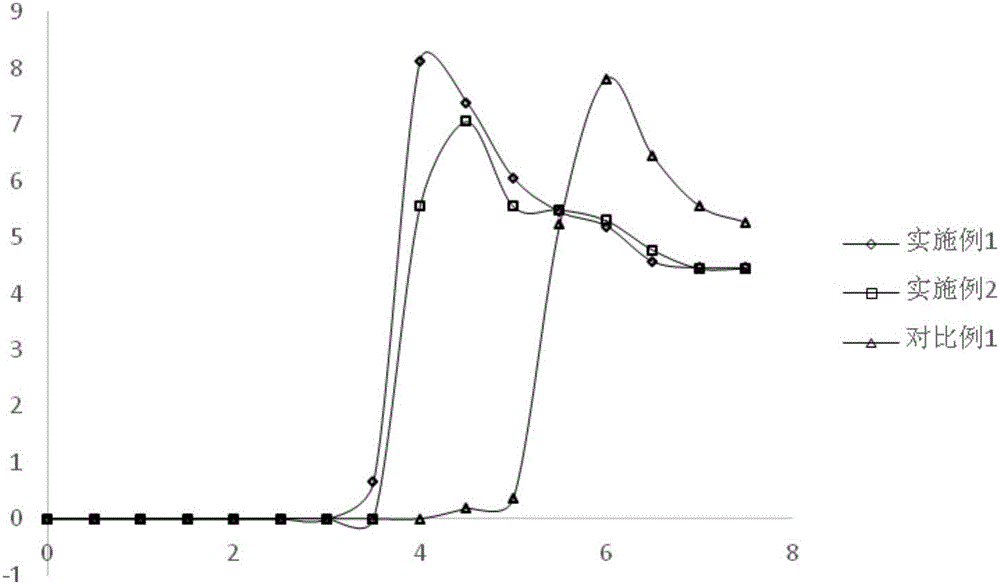
图1示出了根据本发明的实施例的1、实施例2和对比例1中器件的电流密度随电压的变化示意图,其中横坐标为电压(V),纵坐标为电流密度(I/m2);以及
图2示出了根据本发明的实施例的1、实施例2和对比例1中器件的亮度随电压的变化示意图,其中横坐标为电压(V),纵坐标为亮度(Cd/m2)。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
如背景技术所分析的,现有技术的氧化锌纳米晶表面缺陷态严重,氧化锌纳米晶膜层会出现缺陷复合发光的现象,为了解决该问题,本申请提供了一种氧化锌纳米晶、其制备方法、氧化锌纳米晶墨水和电致发光器件。
在本申请一种典型的实施方式中,提供了一种氧化锌纳米晶,氧化锌纳米晶具有表面配体,配体为(硫-磷-烷基),其中,R1、R2和R3各自独立地选自C1~10的烷基中的任意一种。
上述氧化锌纳米晶的表面配体和氧化锌纳米晶形成一层ZnS,既消弱了氧化锌纳米晶主要来自表面Zn的悬挂键导致的缺陷发光,又可以通过调节硫的前驱体种类来方便的调节氧化锌纳米晶的表面配体,进而调节ZnO的电子迁移率。以三丁基硫膦为例,由于氧化锌纳米晶表面的羧酸根表面配体换成了空间位阻更大的三丁基硫膦,增大了相邻氧化锌纳米晶的距离,从而减小了氧化锌纳米晶电子迁移率,在一定程度上缓解了电子和空穴注入的不平衡问题,本申请的氧化锌纳米晶所形成的氧化锌膜的电学性质得到了改进,进而解决了量子点发光二极管的缺陷发光问题。
为了便于合成以及保证所得到的氧化锌纳米晶的结构稳定,优选上述配体为三乙基硫膦、三丁基硫膦(S-TBP)和三辛基硫膦(S-TOP)中的任意一种或多种。进一步优选上述氧化锌纳米晶的表面配体内晶体的粒径为5~20nm,更优选5~10nm,以在使用时形成良好的成膜效果和电子注入效果。
在本申请另一种典型的实施方式中,提供了一种上述氧化锌纳米晶的制备方法,该制备方法包括:步骤S1,采用溶液法制备初始氧化锌纳米晶的溶液,初始氧化锌纳米晶具有羧酸根表面配体;步骤S2,使溶液与硫前驱体在50~200℃下反应,生成氧化锌纳米晶,硫前驱体为S的溶液,其中,R1、R2和R3各自独立地选自C1~10的烷基中的任意一种,且在反应过程中控制反应体系的pH值在7~10之间
利用上述方法,对现有技术的溶液法制备的具有羧酸根表面配体的初始氧化锌纳米晶进行配体修饰,利用硫前驱体S的溶液(即硫溶解于溶液中)和表面的锌离子反应生成一层ZnS,自然地,表面配体换成了与S相连的同时锌上的羧酸根脱落,使溶液呈酸性。为了使步骤S2的反应有效进行,控制反应体系的pH值在7~10之间。可以采用碱金属碱或碱性盐化合物进行pH值的调控,比如采用四甲基氢氧化铵或氢氧化钠。
由于不同的硫前驱体硫的溶液中的R基的长度不同,硫前驱体S的溶液的反应活性不同,为了使硫前驱体得到充分利用,优选上述硫前驱体为S的溶液,其中,R1、R2和R3为C1~4的烷基中的任意一种,步骤S2包括:使溶液与硫前驱体在50~160℃下反应10~60min,生成氧化锌纳米晶。优选硫前驱体为S的溶液,其中,R1、R2和R3为C5~10的烷基中的任意一种,步骤S2包括:使溶液与硫前驱体在160~200℃下反应10~60min,生成氧化锌纳米晶。R基的C链越长,其反应活性越差,因此,根据C链长度确定了不同C链范围的硫前驱体的反应温度,以此来提高硫前驱体的利用率。
此外,为了尽可能使初始氧化锌纳米晶表面的羧酸根配体被替换,优选硫前驱体中的硫与初始氧化锌纳米晶的溶液中初始氧化锌纳米晶摩尔比为10:1~100:1。
上述溶液法制备初始氧化锌纳米晶过程为现有技术,比如:
制备溶液A:将3mmol二水合乙酸锌和30ml DMSO(二甲基亚砜)加入到100ml三口烧瓶中,于30℃水浴环境中加热并磁力搅拌。
制备溶液B:另取一小烧杯,加5mmolTMAH(四甲基氢氧化铵)和10ml乙醇,摇匀混合均匀后用封膜封口。将溶液B逐滴滴加到溶液A中(10min左右完成),然后继续磁力搅拌,在30℃水浴环境下搅拌1小时。将反应结束后的混合溶液经提纯后,得到提纯的初始氧化锌纳米晶,然后将该初始氧化锌纳米晶溶解于溶剂中,形成相应的初始氧化锌纳米晶的溶液,该溶液可以为醇溶液或醚溶液,优选醇溶液为乙醇溶液、乙二醇溶液或丙二醇溶液,优选醚溶液为丙二醇甲醚溶液。将氧化锌纳米晶溶解于醇或者酮中有利于其保存和下一步的反应。
为了进一步提高上述制备方法得到的氧化锌纳米晶的应用价值,优选上述步骤S2包括:步骤S21,使溶液与硫前驱体在50~200℃下反应,得到含有氧化锌纳米晶的产物体系;步骤S22,提纯产物体系中的氧化锌纳米晶。上述提纯的方法可以采用现有技术中常用的量子点提纯方法。
在本申请一种优选的实施例中,上述步骤S22包括:步骤S221,将产物体系与乙酸乙酯混合后进行第一次固液分离,得到下层沉淀物,即得到氧化锌纳米晶,优选采用离心进行上述第一次固液分离。此外,为了进一步减少氧化锌纳米晶表面缺陷,优选上述步骤S22还包括步骤S222,将沉淀物与乙醇、乙醇胺混合后向其中加入乙酸乙酯至混合体系变白色,得到待分离液;步骤S223,对待分离液进行第二次固液分离,得到位于下层的具有乙醇胺配体修饰的氧化锌纳米晶,其中优选采用离心进行第二次固液分离。在上述提纯过程的同时,利用乙醇胺对氧化锌纳米晶的表面进行进一步的修饰,使得乙醇胺可以通过胺基端与氧化锌纳米晶表面的金属锌配位。具体为:由于的空间位阻较大,使氧化锌纳米晶表面一些锌离子没有机会和反应。乙醇胺配体因为其体积小,可以穿过配体层,从而对没有配体的锌离子进一步配位,增加溶解性(乙醇胺作为配体可以提高氧化锌纳米晶在醇类溶液的溶解度)的同时减少氧化锌纳米晶的表面缺陷。
进一步优选上述步骤S221包括:将产物体系与乙酸乙酯以1:2~2:1的体积比混合后进行离心分离,得到下层沉淀物,其中,离心速率为2000~4000rpm,离心时间为1~5min。采用上述操作既能实现对产物体系中的氧化锌纳米晶的充分沉淀,而且不会浪费过多的乙酸乙酯。
此外,优选上述步骤S222还可以包括:将沉淀物与体积比为500:1~2000:1的乙醇和乙醇胺的混合液混合后向其中加入乙酸乙酯至混合体系变白色,得到待分离液;步骤S223中离心的转速为2000~4000rpm、离心时间为1~5min。在上述条件下,利用乙醇对沉淀物充分溶解,然后利用乙醇胺对沉淀物表面修饰后利用乙酸乙酯进行沉淀。
在本申请另一种优选的实施例中,上述步骤S22包括:采用醇和/或酮对产物体系进行洗涤,得到含氧化锌纳米晶的洗涤液;利用乙酸乙酯对洗涤液中的氧化锌纳米晶进行沉淀,得到氧化锌纳米晶,优选其中醇选自乙醇、甲醇、乙二醇和丙二醇中的任意一种或多种,酮为丙酮。利用氧化锌纳米晶在醇和酮中的高溶解性,从而在洗涤过程中,使氧化锌纳米晶和杂质溶解于醇和酮溶液中,加入乙醇胺溶液之后,使氧化锌纳米晶沉淀,杂质被醇和酮带走,实现提纯的目的。
在本申请另一种典型的实施方式中,提供了一种氧化锌纳米晶墨水,包括氧化锌纳米晶和溶剂,该氧化锌纳米晶为上述的任一种氧化锌纳米晶。氧化锌纳米晶以氧化锌纳米晶墨水的形式的存在,一方面有利于其在保证具有稳定发光性能的基础上长期稳定存在,另一方面有利于其应用的工艺。
上述溶剂优选为乙醇、丁醇、庚醇、癸醇、乙二醇、丙二醇、丙二醇醚类、丙二醇酯类中任一种或几种。当溶剂为乙醇或丁醇时,该氧化锌纳米晶墨水可用于旋涂或打印;当溶剂为乙醇、丁醇、庚醇、癸醇、乙二醇、丙二醇、丙二醇醚类或丙二醇酯类时,氧化锌纳米晶墨水可用于打印。其中的丙二醇醚类可以为丙二醇甲醚、丙二醇乙醚、丙二醇丙醚,丙二醇酯类可以为丙二醇甲醚醋酸酯、丙二醇甲酸酯,丙二醇丙酸酯。
在本申请又一种典型的实施方式中,提供了一种电致发光器件,包括电子传输层或/和电子注入层,该电子传输层或/和电子注入层采用上述任一种氧化锌纳米晶墨水制备而成。由于本申请的氧化锌纳米晶的表面得到优化,因此消弱了其缺陷发光,进而当将其应用至电致发光器件中作为电子传输层或电子注入层使用时,缩短了载流子的响应时间,有利于其在电致发光器件中推广应用。
以下将结合实施例和对比例,进一步说明本申请的有益效果。
实施例1
初始ZnO纳米晶的合成:
制备溶液A:将3mmol二水合乙酸锌和30ml DMSO(二甲基亚砜)加入到100ml三口烧瓶中,于30℃水浴环境中加热并磁力搅拌。
制备溶液B:另取一小烧杯,加5mmolTMAH(四甲基氢氧化铵)和10ml乙醇,摇匀混合均匀后用封膜封口。
将溶液B逐滴滴加到溶液A中(10min左右完成),然后继续磁力搅拌,在30℃水浴环境下搅拌1小时。
将反应结束后的混合溶液提纯后得到的初始氧化锌纳米晶溶于乙二醇中,形成初始氧化锌纳米晶的乙二醇溶液,具体过程为:取5ml混合液于离心管中,加入5ml乙酸乙酯,于3000rpm+3min离心,倒掉上清液加入3ml乙二醇溶解。
制备氧化锌纳米晶:
向上述初始氧化锌纳米晶的乙醇溶液中加5mmol S-TBP(三丁基硫膦)溶液在100℃下反应30min,含有所述氧化锌纳米晶的产物体系,其中采用四甲基氢氧化铵控制体系的pH值为9。
提纯氧化锌纳米晶:
取5ml上述产物体系加入到离心管中,然后加入5ml乙酸乙酯,在3000rpm的转速下离心3min;倒掉上清液,然后向离心管中加入1ml乙醇,摇匀溶解;再向离心管中加入20μL乙醇胺,再加2ml左右乙酸乙酯至溶液略微泛白,在3000rpm的转速下离心3min。倒掉上清液,倒掉上清液时倒干净残留的乙酸乙酯。然后,向离心管中加入1ml乙醇,自然溶解后,取出并测UV。
实施例2
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙二醇溶液中加10mmol S-TBP(硫溶解于三丁基膦的溶液)溶液在100℃下反应30min,含有所述氧化锌纳米晶的产物体系。
实施例3
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙二醇溶液中加1mmol S-TBP(三丁基硫膦)溶液在100℃下反应30min,含有所述氧化锌纳米晶的产物体系。
实施例4
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙醇溶液中加5mmol S-TBP(三丁基硫膦)溶液在50℃下反应60min,含有所述氧化锌纳米晶的产物体系。
实施例5
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙二醇溶液中加5mmol S-TBP(三丁基硫膦)溶液在160℃下反应10min,含有所述氧化锌纳米晶的产物体系。
实施例6
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙二醇溶液中加5mmol S-TOP(三辛基硫膦)溶液在160℃下反应60min,含有所述氧化锌纳米晶的产物体系。
实施例7
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的乙二醇溶液中加5mmol S-TOP(三辛基硫膦)溶液在200℃下反应30min,含有所述氧化锌纳米晶的产物体系。
实施例8
与实施例1的不同之处在于,制备氧化锌纳米晶过程中,采用四甲基氢氧化铵控制体系的pH值为10。
实施例9
与实施例1不同之处在于,将反应结束后的混合溶液提纯后得到的初始氧化锌纳米晶溶于丙二醇甲醚中,形成初始氧化锌纳米晶的醚溶液。
实施例10
与实施例1不同之处在于,提纯氧化锌纳米晶的过程,具体为:
取5ml上述产物体系加入到离心管中,然后加入5ml乙酸乙酯,在3000rpm的转速下离心3min;倒掉上清液,然后向离心管中加入1ml乙醇,摇匀溶解;再向离心管中加入10μL乙醇胺,再加2ml左右乙酸乙酯至溶液略微泛白,在3000rpm的转速下离心3min。倒掉上清液,倒掉上清液时要倒干净残留的乙酸乙酯。然后,向离心管中加入1ml乙醇,自然溶解后,取出并测UV,根据需要调整浓度。
实施例11
与实施例1不同之处在于,提纯氧化锌纳米晶的过程,具体为:
取5ml上述产物体系加入到离心管中,然后加入5ml乙酸乙酯,在3000rpm的转速下离心3min;倒掉上清液,然后向离心管中加入1ml乙醇,摇匀溶解;再向离心管中加入50μL乙醇胺,再加2ml左右乙酸乙酯至溶液略微泛白,在3000rpm的转速下离心3min。倒掉上清液,倒掉上清液时要倒干净残留的乙酸乙酯。然后,向离心管中加入1ml乙醇,自然溶解后,取出并测UV,根据需要调整浓度。
实施例12
与实施例1不同之处在于,利用2ml左右的乙醇洗涤产物体系,得到氧化锌纳米晶的乙醇溶液。
对比例1
以实施例1得到的初始氧化锌纳米晶的作为对比例1的样品。
对比例2
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的丙二醇溶液中加5mmol S-TOP(三辛基硫膦)溶液在100℃下反应30min,得到对比例2的产物。
对比例3
与实施例1的不同之处在于,制备氧化锌纳米晶:向初始氧化锌纳米晶的丙二醇溶液中加5mmol S-TBP(三丁基硫膦)溶液在200℃下反应30min,得到对比例3的产物。
将实施例1至11和对比例1的浓度为60mg/ml氧化锌纳米晶的乙醇溶液应用于量子点电致发光器件中,具体应用如下:
1)对厚度为200nm的阳极ITO(氧化铟锡)进行清洗处理。
用乙醇、去离子水和丙酮分别超声清洗处理10分钟,然后用N2吹干附着于玻璃表面的液体,并经过臭氧-紫外处理10分钟,以清除ITO表面的杂质,得到清洁ITO透明导电玻璃。
2)制作第一功能层。
在空气环境中,在清洁ITO透明导电玻璃上以4000转/分钟的转速旋涂PEDOT:PSS(聚对苯乙烯磺酸溶液),时间为50秒;旋涂完成后在空气中150℃下退火处理30分钟;再将其转移至氮气环境的手套箱中,130℃退火20分钟,最终在ITO表面形成一层PEDOT:PSS层,即形成空穴注入层。然后在PEDOT:PSS层上以2000转/分钟的转速旋涂聚乙烯基咔唑poly(N—vinylcarbazole)(PVK)的氯苯溶液(浓度为8mg/ml),旋涂时间为45秒;旋涂完成后在手套箱中150℃退火30分钟形成PVK空穴传输层。
3)制作量子点发光层。
量子点为CdSe/CdS核壳结构,发射波长为630nm的红光量子点,分散在正辛烷中,浓度为30mg/ml,转速为2000转/分钟,旋涂时间为45秒。
4)制作第二功能层。
在第二电荷调节层上再旋涂一层实施例1至12和对比例1至3中一种的氧化锌纳米晶的乙醇溶液,转速为2000转/分钟,旋涂时间为45秒。
5)制作阴极。
将旋涂完成的器件置于真空蒸镀仓内,蒸镀阴极银电极,厚度为100nm,得到量子点电致发光器件。
器件测试方法:采用UV3600荧光光谱仪测量发射峰对应的波长(峰值波长)进行测试,采用Keithley2400测定量子点发光器件的电流密度-电压曲线,采用积分球(FOIS-1)结合海洋光学的光谱仪(QE-6500)测定量子点发光器件的亮度,根据测定得到的电流密度与亮度计算量子点发光器件的外量子效率,外量子效率表征在观测方向上发光器件发出的光子数与注入器件的电子数之间的比值,是表征器发光器件发光效率的重要参数,外量子效率越高,说明器件的发光效率越高。
表1
图1和图2分别是对比例1、实施例1、实施例2的外量子效率及亮度-电压曲线,表中的对比例1、实施例1、实施例2的外量子效率分别对应于图2中对比例1、实施例1、实施例2的外量子效率曲线的最高点的数值。可以看到经过S-TBP和S-TOP修饰的ZnO作电子传输层后,器件的外量子效率有所提升。很明显地,实施例1在更低的电压达到了最高外量子效率,说明S-TBP修饰可以很好的去除氧化锌纳米晶的表面缺陷,提高了能量的利用率。
另外,实施例1的氧化锌纳米晶所制备得到的器件测试得到的外量子效率为8.14%,对比例1的初始氧化锌纳米晶所制备得到的器件测试得到的外量子效率为7.06%,实施例2所制备的器件测试得到的外量子效率为8.36%,说明交换后的带S-P-R3配体的氧化锌纳米晶要优于未交换前的初始氧化锌纳米晶。
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
上述氧化锌纳米晶的表面配体和氧化锌纳米晶形成一层ZnS,既消弱了氧化锌纳米晶主要来自其表面Zn的悬挂键导致的缺陷发光,又可以通过调节硫的前驱体种类来方便的调节氧化锌纳米晶的表面配体,进而调节氧化锌纳米晶的电子迁移率。以三丁基硫膦为例,由于氧化锌纳米晶表面的羧酸根表面配体换成了空间位阻更大的三丁基硫膦,增大了相邻氧化锌纳米晶的距离,从而减小了氧化锌纳米晶电子迁移率,在一定程度上缓解了电子和空穴注入的不平衡问题,本申请的氧化锌纳米晶所形成的氧化锌纳米晶体膜的电学性质得到了改进,进而解决了量子点发光二极管的缺陷发光问题。
利用上述方法,对现有技术的溶液法制备的具有羧酸根表面配体的初始氧化锌纳米晶进行配体修饰,利用硫前驱体S的溶液和表面的锌离子反应生成一层ZnS,自然地,ZnS表面配体换成了与S相连的也即氧化锌纳米晶体的表面配体
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!