用于烃油的加氢裂化催化剂、用于生产加氢裂化催化剂的方法以及用加氢裂化催化剂加氢裂化烃油的方法与流程
用于烃油的加氢裂化催化剂、用于生产加氢裂化催化剂的方法以及用加氢裂化催化剂加氢裂化烃油的方法发明
背景技术:
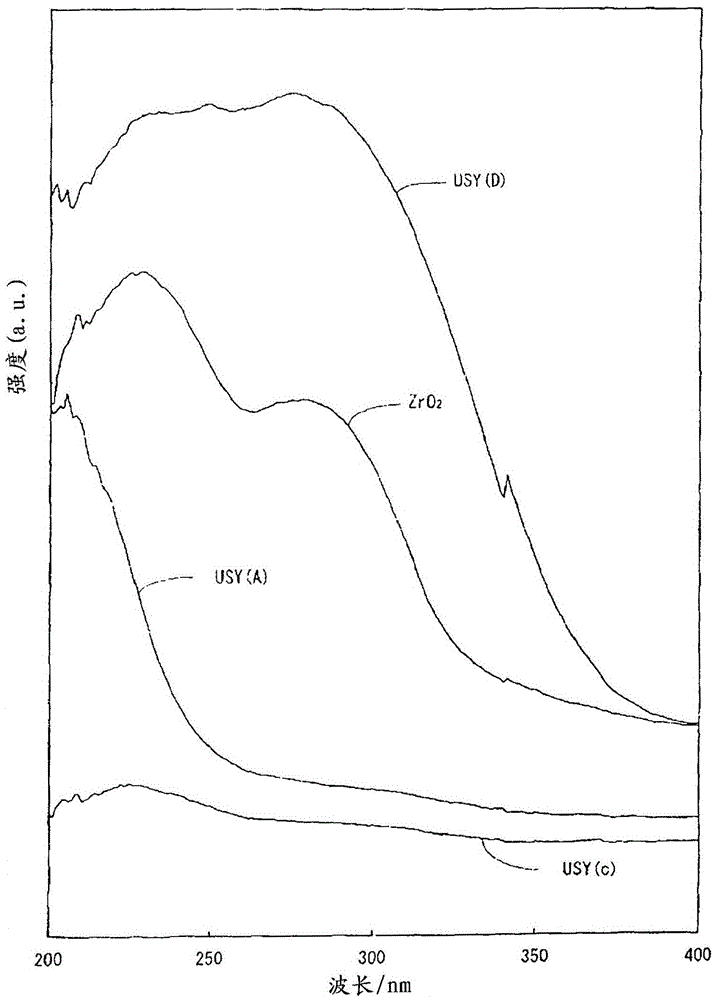
领域本发明尤其涉及用于烃油的加氢裂化催化剂,所述催化剂能以高收率从重质烃,诸如真空瓦斯油(下文也称为“VGO”)和脱沥青油(以下称为“DAO”)生产中间馏分(煤油和瓦斯油;煤油-瓦斯油)。相关领域描述迄今为止,包含沸石的加氢处理催化剂已用于处理残油(例如参见日本未审专利申请公开第2000-334305号、第2002-255537号和第2003-226519号),所述沸石中钛和/或锆负载在(结合到)中孔上。例如,日本未审专利申请公开第2000-334305号公开了一种包含负载在催化剂载体上的加氢金属的氢化催化剂,该载体由沸石组成,在所述沸石中,由二氧化钛或氧化锆的钛族金属氧化物组成的超细颗粒结合到中孔的内表面,和该载体具有在沸石中的0.01-0.1的铝硅原子比,即[Al]/[Si](换言之,SiO2与Al2O3的摩尔比(以下称为“SiO2/Al2O3摩尔比”或二氧化硅-氧化铝比)为20-200)。其中描述了,结合了由钛族金属氧化物组成的超细颗粒的沸石如下制备:将具有中孔的原料沸石与pH为0.8-2的二氧化钛或氧化锆的钛族金属盐的水溶液接触,用水洗涤沸石,干燥得到的沸石,和在400℃-600℃下烧制干燥的沸石。日本未审专利申请公开第2002-255537号公开了一种具有高的中孔含量的沸石,该沸石的铝硅原子比即[Al]/[Si]为0.01-0.2(换言之,二氧化硅-氧化铝比为10-200),孔径为的中孔的体积百分比为30-50%,中孔体积为0.14cc/g或以上,四配位铝原子占总铝原子的份额为25原子百分比或以上,其中不易还原的二氧化钛和/或氧化锆的金属氧化物超细颗粒结合到上述沸石的中孔的内表面,以及公开了包括加氢金属的加氢处理催化剂,该加氢金属负载在由上述沸石组成的催化剂载体上。具有高的中孔含量的沸石如下制备:将原料沸石与pH为0.8-2的强酸性水溶液接触,在50-200℃下干燥沸石,和在350℃-600℃下烧制干燥的沸石。其中描述了,由此可以制备其中将金属氧化物超细颗粒结合到(负载在)孔内表面的沸石。日本未审专利申请公开第2003-226519号公开了用于烃油的加氢处理催化剂,该加氢处理催化剂包括改性沸石,其中晶格常数为或以上和或以下的八面沸石包含元素周期表第4族中的金属元素(钛、锆或铪),基于元素金属计,改性沸石的金属元素含量为0.1-10重量%,Al/Si原子比为0.01-0.1(换言之,二氧化硅-氧化铝比为20-200),该加氢处理催化剂还包含加氢金属。其中描述了,改性沸石如下制备:在酸性条件下将晶格常数为的八面沸石与包含元素周期表第4族中的元素的水溶性化合物的水溶液接触。然而,在这些加氢处理催化剂中,中孔被负载的金属堵塞,因此这些催化剂在某些情况下不适用于重质烃油,诸如VGO和DAO的加氢处理(或加氢裂化)。如WO2007/032232中所公开,已经开发了包括Y-型沸石作为载体的加氢处理催化剂,该Y-型沸石包含结合到沸石骨架中的钛原子(换言之,Y-型沸石中构成骨架的铝原子被钛原子所取代)。上述沸石可由以下制备:用pH为1.5或以下的含钛的酸性水溶液处理Y-型沸石,接着过滤、洗涤和干燥。由此,可以使沸石包含结合到沸石骨架结构中而不阻塞中孔的钛原子。其中描述了,当将包括上述沸石作为载体的加氢处理催化剂应用于重质烃油的加氢处理时,由于重质烃油易于扩散到中孔中,提高了中间馏分的收率。发明简述然而,包括作为载体的其中构成沸石骨架的铝原子的一部分被钛原子替换的沸石的加氢处理催化剂具有极高的反应性(分解活性)并且过度地分解煤油-瓦斯油,因而不利地降低了中间馏分的收率。鉴于前述情况得到了本发明。本发明的目的是提供用于烃油的加氢裂化催化剂,该加氢裂化催化剂能够以高收率提供中间馏分,还提供生产该加氢裂化催化剂的方法和使用该加氢裂化催化剂的加氢裂化方法。按照上述目的,根据本发明的用于烃油的加氢裂化催化剂是包括负载在载体上的加氢金属组分的用于烃油的加氢裂化催化剂,该载体包含超稳定Y-型沸石,其中上述超稳定Y-型沸石是骨架取代沸石(以下称为骨架取代沸石-1),在该沸石中构成其沸石骨架的铝原子的一部分被锆原子和/或铪原子取代。在根据本发明的用于烃油的加氢裂化催化剂中,以氧化物计0.1-5质量%的锆原子和/或铪原子优选包含在上述骨架取代沸石-1中。在根据本发明的用于烃油的加氢裂化催化剂中,骨架取代沸石-1中进一步优选包含钛原子。在根据本发明的用于烃油的加氢裂化催化剂中,骨架取代沸石-1中构成沸石骨架的铝原子的一部分优选被钛原子取代。在根据本发明的用于烃油的加氢裂化催化剂中,以氧化物计0.1-5质量%的钛原子优选包含在骨架取代沸石-1中。在根据本发明的用于烃油的加氢裂化催化剂中,上述载体优选包含骨架取代沸石-1和除上述骨架取代沸石-1之外的无机氧化物。在根据本发明的用于烃油的加氢裂化催化剂中,上述无机氧化物优选为铝或二氧化硅-氧化铝。在根据本发明的用于烃油的加氢裂化催化剂中,上述载体进一步包含优选骨架取代沸石(以下称为骨架取代沸石-2),其中构成超稳定Y-型沸石骨架的铝原子的一部分仅被钛原子取代。在根据本发明的用于烃油的加氢裂化催化剂中,当上述载体包含骨架取代沸石-2时,上述骨架取代沸石-2优选包含以氧化物计0.1-5质量%的钛原子。在根据本发明的用于烃油的加氢裂化催化剂中,上述载体优选包含骨架取代沸石-1、骨架取代沸石-2和除上述骨架取代沸石-1和上述骨架取代沸石-2之外的无机氧化物。在根据本发明的用于烃油的加氢裂化催化剂中,载体中包含的骨架取代沸石-1优选具有下列性质(a)-(c):(a)晶格常数为2.430-2.450nm,(b)比表面积为600-900m2/g和(c)SiO2与Al2O3的摩尔比为20-100。在根据本发明的用于烃油的加氢裂化催化剂中,该催化剂的比表面积优选为200-450m2/g;直径为或以下的孔的体积优选为0.40-0.75ml/g;和加氢金属组分的负载量优选为0.01-40质量%。本发明中用于生产用于烃油的加氢裂化催化剂的方法为生产用于烃油的加氢裂化催化剂的方法,包括将加氢金属组分负载在包含骨架取代沸石-1的载体上,在所述骨架取代沸石-1中构成超稳定Y-型沸石骨架的铝原子的一部分被锆原子和/或铪原子取代,以及上述骨架取代沸石-1如此获得:烧制超稳定Y-型沸石,该超稳定Y-型沸石的晶格常数为2.430nm或以上和2.450nm或以下,比表面积为600-900m2/g,和在500-700℃下SiO2与Al2O3的摩尔比为20-100,由上述经烧制的超稳定Y-型沸石制备液/固质量比为5-15的悬浮液,向其中添加无机酸或有机酸以使上述悬浮液的pH为1.0-2.0,随后添加锆化合物和/或铪化合物并将它们混合,然后中和该悬浮液。在本发明中的用于生产用于烃油的加氢裂化催化剂的另一方法中,将如此获得的沸石用作骨架取代沸石-1:烧制超稳定Y-型沸石,该超稳定Y-型沸石的晶格常数为2.430nm或以上和2.450nm或以下,比表面积为600-900m2/g,在500-700℃下SiO2与Al2O3的摩尔比为20-100,由上述经烧制的超稳定Y-型沸石制备液/固质量比为5-15的悬浮液,向其中添加无机酸或有机酸以使上述悬浮液的pH为1.0-2.0,随后添加锆化合物和/或铪化合物并将它们混合,然后中和该混合溶液。按照上述目的,根据本发明的第三方面,用于加氢裂化烃油的方法包括使用上述加氢裂化催化剂加氢裂化烃油。优选地,根据本发明第三方面的用于加氢裂化烃油的方法进一步包括用加氢裂化催化剂填充加氢裂化装置的反应容器,该加氢裂化装置为连续反应器,和在氢气存在下在反应器温度为300℃-500℃、氢气压力为4-30MPa、液体时空速(LHSV)为0.1-10h-1和氢气/油比为500-2500Nm3/m3的下处理沸点为375℃-816℃(707-1500℉)的烃油。优选地,根据本发明第三方面的用于加氢裂化烃油的方法进一步包括用加氢裂化催化剂填充加氢裂化装置的反应容器,该加氢裂化装置为连续反应器,和在氢气存在下在反应器温度为330℃-450℃、氢气压力为7-15MPa、液体时空速(LHSV)为0.2-1.5h-1和氢气/油比为1000-2000Nm3/m3的下处理沸点为375℃-650℃(707-1200℉)的烃油以提供煤油-瓦斯油。在根据本发明的用于加氢裂化烃油的方法中,上述连续反应器优选为连续反应器,该连续反应器选自搅拌浴型反应器、沸腾床型反应器、装有折流板的淤浆浴型反应器、固定床型反应器、旋转管型反应器和淤浆床型反应器。在根据本发明的用于加氢裂化烃油的方法中,上述烃油优选包含得自(1)原油、(2)合成原油、(3)沥青、(4)油砂、(5)页岩油或(6)煤馏油的精制油。在根据本发明的用于加氢裂化烃油的方法中,上述烃油包含得自原油、合成原油、沥青、油砂、页岩油或煤馏油的精制油,上述精制油优选为a)真空瓦斯油(VGO),b)得自溶剂脱沥青过程的脱沥青油(DAO)或脱金属油,c)得自焦化过程的轻质焦化瓦斯油或重质焦化瓦斯油,d)得自流化床催化裂化(FCC)过程的循环油或者e)得自减黏裂化过程的瓦斯油的任一种。根据本发明的用于的烃油加氢裂化催化剂的特征在于在用于烃油的加氢裂化催化剂中包括负载在包含超稳定Y-型沸石的载体上的加氢金属组分,上述超稳定Y-型沸石为骨架取代沸石-1,其中构成其骨架的铝原子的一部分被锆原子和/或铪原子取代。因此,本发明的加氢裂化催化剂与包括负载了钛微细颗粒或锆微细颗粒的沸石载体的常规加氢裂化催化剂相比,易于将重质烃如VGO、DAO等扩散到它的中孔中,和增强了烃油的裂化活性从而可能以高收率获得中间馏分。另外,本发明的加氢裂化催化剂与常规加氢裂化催化剂相比烃油的裂化活性稍低,所述常规加氢裂化催化剂包括负载在包括骨架取代沸石的载体上的加氢金属组分,所述骨架取代沸石中构成Y-型沸石骨架的铝原子的一部分被钛原子取代,但是本发明的加氢裂化催化剂抑制了煤油和瓦斯油的过度裂化反应,因此可以以高收率获得中间馏分。而且,根据本发明的用于烃油的加氢裂化催化剂上活性位点的数量增多,因此具有高加氢裂化活性。附图简述图1为USY(A)、USY(c)和ZrO2的UV-vis-NIR光谱。图2为USY(A)、USY(D)和ZrO2的UV-vis-NIR光谱。图3为USY(A)和USY(H)的UV-vis-NIR光谱。在所有USY(A)、USY(H)和USY(D)中,在208nm波长附近观察到峰,正如ZrO2那样。USY(c)为用于USY(A)、USY(G)和USY(E)的原料的超稳定Y-型沸石的光谱。图4为USY(A)、USY(G)、USY(E)和USY(c)的FT-IR光谱。USY(A):在960cm-1波长附近观察到基于Si-O-Zr的峰。这显示USY(A)为骨架取代沸石,其中构成USY(A)沸石骨架的Al原子的一部分被Zr原子取代。USY(G):在960cm-1波长附近观察到基于Si-O-Ti的峰。这显示USY(G)为骨架取代沸石,其中构成USY(A)沸石骨架的Al原子的一部分被Ti原子取代。USY(E):在960cm-1波长附近观察到基于Si-O-Zr和Si-O-Ti的峰。这显示USY(E)为骨架取代沸石,其中构成USY(A)沸石骨架的Al原子的一部分被Zr原子和/或Ti原子取代。USY(c):USY(c)为用于USY(A)、USY(G)和USY(E)的原料的超稳定Y-型沸石。优选实施方式的描述用于烃油的加氢裂化催化剂根据本发明的用于烃油的加氢裂化催化剂包括负载在包含骨架取代沸石-1的载体上的加氢金属组分,所述骨架取代沸石-1包含锆原子和/或铪原子部分构成的超稳定Y-型沸石骨架(下文也称为“USY”)。在本说明书中,“根据本发明的用于烃油的加氢裂化催化剂”会被称为“根据本发明的加氢裂化催化剂”,以及“根据本发明的用于生产用于烃油的加氢裂化催化剂的方法”会被称为“根据本发明的用于生产加氢裂化催化剂的方法”。以下会详细解释根据本发明的加氢裂化催化剂。(1)骨架取代沸石-1(结构)本发明的骨架取代沸石-1为超稳定Y-型沸石,其中硅原子和铝原子形成沸石骨架,和其中铝原子的一部分被锆原子和/或铪原子取代(以下将其中形成沸石骨架的铝原子的一部分仅被锆原子取代的骨架取代沸石-1称为“锆-取代沸石”或“Zr-USY”;将其中形成骨架取代沸石-1的沸石骨架的铝原子的一部分仅被铪原子取代的骨架取代沸石-1称为“铪-取代沸石”或“Hf-USY”;并且类似地,将其中形成沸石骨架的铝原子的一部分仅被锆原子和铪原子取代的骨架取代沸石-1称为“锆·铪-取代沸石”或“Zr·Hf-USY”)。取代铝原子形成超稳定Y-型沸石骨架的锆原子和/或铪原子作为超稳定Y-型沸石骨架的组成部分。在该方面,本发明中的“取代”不同于“负载”,负载是将锆原子和/或铪原子或者它们的颗粒附着在超稳定Y-型沸石骨架的外面,并且本发明的“取代”也不同于上述专利文献2(日本未审专利申请公开第2002-255537号)的权利要求3中限定的“结合”。在根据本发明的骨架取代沸石-1中,锆原子和/或铪原子可以如专利文献2的权利要求3中所限定,以例如氧化物的形式任选“负载”其上或与其“结合”。可通过例如紫外线、可见光和近红外线分光光度法(UV-Vis-NIR)、傅里叶变换红外光谱法(FT-IR)或核磁共振光谱测定法(NMR)来证实取代确已发生。注意,在β-沸石骨架已被锆原子取代的骨架取代沸石中,已知指示锆原子存在的UV光谱显示在约200-约220nm范围内(例如参见“B.Raksheetal.,JournalofCatalysis188,252,1999”的图3)。在本发明的骨架取代沸石-1中,形成其沸石骨架的铝原子的一部分必须被(I)锆原子、(II)铪原子或(III)氧化锆和铪原子取代。本发明的骨架取代沸石-1优选包含0.1-5质量%,更优选为0.2-4质量%的锆原子和/或铪原子,以基于骨架取代沸石-1的氧化物(即“ZrO2”和“HfO2”)计。进一步地,更优选建议值为0.3-3质量%。在这方面,锆原子和/或铪原子的含量范围(基于氧化物计)包括取代形成沸石骨架的铝原子的锆原子和/或铪原子以及未取代上述铝原子的锆原子和/或铪原子的全部的含量。当将通过使用上述骨架取代沸石-1作为载体来制备的加氢裂化催化剂应用于加氢裂化反应器时,以基于骨架取代沸石-1质量的氧化物计,骨架取代沸石-1中锆原子和/或铪原子含量少于0.1质量%不导致用于加氢裂化反应器的有效量的固体酸,因此容易引起烃油加氢裂化反应器活性的降低。类似地,以基于骨架取代沸石-1的氧化物计,锆原子和/或铪原子含量超过5质量%不导致用于烃油加氢裂化反应器的有效孔体积,因此容易引起烃油加氢裂化反应器活性的降低。当本发明的骨架取代沸石-1包含上述锆原子和铪原子时,锆原子与铪原子的质量比(以氧化物计)应当没有特别限制。可采用例如X射线荧光分析仪、高频等离子体发射光谱仪、原子吸收光谱仪等测量骨架取代沸石-1的锆原子和/或铪原子含量。在上述骨架取代沸石-1中,锆原子和/或铪原子可以如专利文献2的权利要求3所限定,以例如氧化物的形式任选“负载”其上或与其“结合”。在这种情况下,如上所述,锆原子可以以氧化锆颗粒的形式负载或结合。如上所述,铪原子也可以以氧化铪颗粒的形式负载或结合。当存在上述颗粒时,其粒径优选为50nm或以下。超过50nm的各上述锆颗粒和/或铪颗粒的粒径在某些情况下不导致用于加氢处理反应器的有效孔体积并导致孔的堵塞。因此,采用包括上述沸石的加氢裂化催化剂的氢化和脱氢活性趋向下降。可通过使用扫描电子显微镜(SEM)拍摄的照片来测量各上述锆颗粒和铪颗粒的粒径。本发明的骨架取代沸石-1除锆原子和/或铪原子之外可以包含钛原子,并且上述钛原子更优选的包含方式为钛原子部分地取代形成沸石骨架的铝原子的一部分。具体而言,上述骨架取代沸石-1中包含的钛原子份额优选为0.1-5质量%,更优选为0.5-4质量%,以基于骨架取代沸石-1的氧化物(即TiO2)计。同样,进一步优选建议0.6-3质量%的份额。在这方面,如果骨架取代沸石-1中上述钛原子以氧化物计的含量少于0.1质量%,当将通过使用上述骨架取代沸石-1作为载体来制备的加氢裂化催化剂应用于加氢裂化反应器时,未获得对加氢裂化反应器而言有效的固体酸量,因此加氢裂化反应器中烃油的活性趋向降低。类似地,如果骨架取代沸石-1中钛原子以氧化物计的含量超过5质量%,当将通过使用上述骨架取代沸石-1作为载体来制备的加氢裂化催化剂应用于加氢裂化反应器时,未获得对加氢裂化反应器而言有效的孔体积,因此加氢反应器和加氢裂化反应器中烃油的活性趋向降低。可以通过例如X射线荧光分析仪、高频等离子体发射光谱仪、原子吸收光谱仪等测量骨架取代沸石-1中钛原子的含量。(2)骨架取代沸石-2(结构)在根据本发明的加氢裂化催化剂中,除上述骨架取代沸石-1外,可以包含其中形成超稳定Y-型沸石的铝原子的一部分仅被钛原子和/或无机酸(仅限于用于骨架取代沸石-1的那些不相应的无机酸)取代的骨架取代沸石(以下称为“骨架取代沸石-2”)作为载体。未取代上述铝原子的钛原子可包含在骨架取代沸石-2(“骨架取代沸石-2”被称为“钛-取代沸石”或“Ti-USY”)中。可通过例如WO2007/032232(专利文献4)所述的方法来制备骨架取代沸石-2。上述骨架取代沸石-2优选包含0.1-5质量%,更优选为0.5-4质量%的钛原子以基于骨架取代沸石-2的氧化物(即“TiO2”)计。进一步优选建议0.6-3质量%的含量。上述钛原子的含量范围(基于氧化物计)包括取代形成沸石骨架的铝原子的钛原子和未取代上述铝原子的钛原子的全部的含量。在这方面,以氧化物计各骨架取代沸石-2的钛原子的含量少于0.1质量%不导致用于加氢裂化反应器有效量的固体酸,因此容易引起加氢裂化反应器中烃油活性的降低。含量超过5质量%不导致用于加氢裂化反应器的有效的孔体积,因此容易引起加氢反应器和加氢裂化反应器中烃油活性的降低。采用例如X射线荧光分析仪、高频等离子体发射光谱仪、原子吸收光谱仪等测定骨架取代沸石-2的钛原子的含量。在这方面,骨架取代沸石-2的晶格常数、比表面积、二氧化硅-氧化铝比、结晶度和孔径为或以下的孔的体积优选与骨架取代沸石-1的范围相同。(3)骨架取代沸石-1的特性本发明的骨架取代沸石-1的晶格常数、比表面积、SiO2与Al2O3的摩尔比即二氧化硅-氧化铝比优选处于预定范围内。(a)晶格常数(UD)本发明的骨架取代沸石-1的晶格常数优选为2.430-2.450nm,更优选为2.435-2.445nm。骨架取代沸石的晶格常数小于2.430nm容易因为沸石骨架结构中的SiO2/Al2O3摩尔比高和用作烃分解活性位点的固体酸位数量少而引起通过使用骨架取代沸石-1作为载体来制备的加氢裂化催化剂活性的降低。骨架取代沸石-1的晶格常数超过2.450nm导致骨架取代沸石-1的晶体结构在加氢裂化反应器时由于骨架取代沸石-1的耐热性低而受破坏,因此容易引起通过使用骨架取代沸石-1作为载体来制备的加氢裂化催化剂活性的降低。上述骨架取代沸石-2的晶格常数也优选为2.430-2.450nm,更优选为2.435-2.445nm。优选上述晶格常数范围的原因与骨架取代沸石-1的情形相同。在此,可以通过参照ASTM法来测量晶格常数:用硅(Si)作为主参照材料来确定氧化钛(锐钛矿)的(111)面的Kα角度。使用氧化钛作为第二参照材料来确定Y沸石的(533)和(642)面的X射线衍射峰。(b)比表面积(SA):本发明的骨架取代沸石-1的比表面积优选为600-900m2/g,更优选为650-800m2/g。该比表面积是通过使用氮气吸附的BET法确定的值。骨架取代沸石-1的比表面积少于600m2/g在某些情况下导致用于加氢处理反应器有效的固体酸位数量减少,由此使得通过使用上述骨架取代沸石作为载体来制备的加氢裂化催化剂的活性不令人满意。比表面积超过900m2/g的骨架取代沸石难于生产。骨架取代沸石-2的比表面积也优选为600-900m2/g,更优选为650-800m2/g。优选上述比表面积范围的原因与骨架取代沸石-1的情形相同。(c)SiO2与Al2O3的摩尔比(二氧化硅-氧化铝比):本发明的骨架取代沸石-1的SiO2与Al2O3的摩尔比(二氧化硅-氧化铝比)优选为20-100,更优选为25-80。骨架取代沸石-1的二氧化硅-氧化铝比小于20不导致对于加氢处理反应器的有效孔体积,因此容易引起采用通过使用骨架取代沸石作为载体来制备的加氢裂化催化剂的氢化和加氢裂化反应的活性降低。骨架取代沸石-1的二氧化硅-氧化铝比超过100则因为对于加氢处理反应器有效的固体酸位数量少而容易引起采用通过使用骨架取代沸石来制备的加氢裂化催化剂的分解反应器中的活性降低。骨架取代沸石-2的二氧化硅-氧化铝比也优选为20-100,更优选为25-80。优选上述二氧化硅-氧化铝比范围的原因与骨架取代沸石-1的情形相同。(d)结晶度:骨架取代沸石-1的结晶度优选为80%或以上。结晶度少于80%则不提供包括骨架取代沸石作为载体的加氢裂化催化剂的期望的效果。骨架取代沸石-1的结晶度可取地为100%-130%。骨架取代沸石-2的结晶度趋向与骨架取代沸石-1的情形相同。在这方面,结晶度如下确定:确定通过X射线衍射测量的骨架取代沸石的(331)、(511)、(440)、(533)、(642)和(555)面的峰的总高度(H)。确定商业上可获得的Y沸石(SK-40,UnionCarbideCorporation制造)相同面的峰的总高度(H0)作为参照。使用下式(1)来确定结晶度:结晶度(%)=H/H0×100(1)骨架取代沸石-2的结晶度也优选为80%或以上。优选上述结晶度范围的原因与骨架取代沸石-1的情形相同。(4)生产骨架取代沸石-1的方法本发明的骨架取代沸石-1可通过例如如下所述的方法生产。骨架取代沸石-1通过如下生产:在500℃-700℃下烧制超稳定Y-型沸石,该超稳定Y-型沸石的晶格常数为2.430-2.450nm,比表面积为600-900m2/g,SiO2与Al2O3的摩尔比为20-100,形成包含经烧制的超稳定Y-型沸石的悬浮体,该悬浮体的液/固质量比为5-15,添加无机酸或有机酸以使上述悬浮体的pH为1.0-2.0,随后添加包含锆化合物和/或铪化合物的溶液并将它们混合,并且采用例如氨水中和所述溶液,使得混合溶液的pH为约7。上述生产方法将在下文详细描述。a)超稳定Y-型沸石将超稳定Y-型沸石用作本发明的骨架取代沸石-1的原料之一。超稳定Y-型沸石是公知的,用于其生产的方法应当没有特别限制。本发明的超稳定Y-型沸石是指晶格常数(UD)为2.430nm或以上和2.450nm或以下、比表面积为600-900m2/g和SiO2与Al2O3的摩尔比(二氧化硅-氧化铝比)为20-100的沸石。在用于上述超稳定Y-型沸石的生产方法中,使通过常规方法合成的Y-型沸石(Na-Y)经受通过常规方法将钠离子用铵离子交换(例如,将Y-型沸石分散到水中以制备分散体,向其中添加硫酸铵,然后用水洗涤固体物质,接着将其用温度为40-80℃的硫酸铵水溶液洗涤,随后将其用40-95℃的水洗涤,然后将其在100-180℃下干燥30分钟)以获得铵-交换的Y-型沸石(NH4-50-70Y),其中Y-型沸石中包含的50-70%的钠被NH4取代。随后,通过将上述铵-交换的Y-型沸石(NH4-50-70Y)在500-800℃于饱和蒸汽气氛中煅烧10分钟-10小时来制备氢型Y-型沸石(HY)。通过将上面获得的氢型Y-型沸石分散到40-95℃的水中以制备分散体,再向其中添加硫酸铵,然后将分散体在40-95℃搅拌10分钟-3小时,再将固体物质用40-95℃的水洗涤,接着将其用40-95℃的硫酸铵水溶液洗涤,随后将其用40-80℃的水洗涤,然后将其在100-180℃干燥30分钟-30小时可以获得铵-交换的Y-型沸石(NH4-80-97Y),其中初始的Y-型沸石(Na-Y)中包含的80-97%的钠被NH4取代。在这方面,最终的铵离子交换率优选为90%或以上。将由此获得的铵-交换的Y-型沸石(NH4-80-97Y)在500-700℃于例如饱和蒸汽气氛中煅烧10分钟-10小时,由此能够制得的是晶格常数(UD)为2.430nm或以上和2.450nm或以下、比表面积为600-900m2/g和SiO2与Al2O3的摩尔比(二氧化硅-氧化铝比)为20-100的超稳定Y-型沸石(USY)。对于获得期望的骨架取代沸石-1而言重要的是将超稳定Y-型沸石的晶格常数控制在2.430-2.450nm。在根据本发明的用于生产加氢裂化催化剂的方法中,可以将骨架外的铝(非形成沸石骨架的铝原子)从作为原料的上述超稳定Y-型沸石中除去以获得晶格常数为2.430-2.450nm的超稳定Y-型沸石。可通过例如下述方法来除去骨架外的铝:将上述超稳定Y-型沸石分散到40-95℃的温水中以制备分散体,将硫酸添加到上述分散体中并将其搅拌10分钟-3小时,同时将温度保持在40-95℃,从而将骨架外的铝溶解。硫酸的添加量应当没有特别限制,只要该量可以将骨架外铝溶解到期望的水平即可。在溶解骨架外的铝之后,过滤分散体,用40-95℃的净化水洗涤过滤器上的残留物并在100-180℃干燥3-30小时,从而可以获得除去了骨架外的铝的超稳定Y-型沸石。在根据本发明的用于生产加氢裂化催化剂的方法中,将作为原料的超稳定Y-型沸石在500℃-700℃,优选550℃-650℃煅烧。煅烧时间应当没有特别限制,只要获得目标骨架取代沸石-1,并且将其在例如30分钟-10小时的范围内煅烧即可。如果超稳定Y-型沸石的煅烧温度低于500℃,当在后续步骤通过锆原子、铪原子或钛原子进行骨架取代处理时,与煅烧在500℃-700℃下进行的情况相比,锆原子、铪原子和钛原子的骨架取代量趋向减少。如果煅烧温度超过700℃,超稳定Y-型沸石的比表面积降低,和当在后续步骤通过锆原子、铪原子或钛原子进行骨架取代处理时,锆原子、铪原子和钛原子的骨架取代量减少,由此锆原子、铪原子和钛原子变得以特殊形式存在。关于超稳定Y-型沸石的煅烧气氛,优选在空气中进行煅烧。将经煅烧的超稳定Y-型沸石悬浮在温度为约20℃-约30℃的水中以形成悬浮体。至于超稳定Y-型沸石悬浮体的浓度,液/固质量比优选为5-15,更优选地,建议质量比为8-12。接着,将无机酸或有机酸添加到其中,从而将上述悬浮体的pH控制在1.0-2.0,随后添加包含锆化合物和/或铪化合物的溶液并混合。然后,将混合溶液中和(pH为7.0-7.5)并可取地在80-180℃干燥,由此可以获得上述骨架取代沸石-1。可以将硫酸、硝酸、盐酸等作为上述无机酸使用,其中特别优选硫酸、盐酸等。另外,可以适宜地将羧酸用作上述有机酸。无机酸或有机酸的使用量应当没有特别限制,只要可以将悬浮体的pH控制在1.0-2.0,和基于超稳定Y-型沸石中Al2O3的量例如0.5-4.0倍的摩尔量,优选为0.7-3.5倍的摩尔量,但并不限制在上述范围内。上述锆化合物的实例包括硫酸锆、硝酸锆、氯化锆等。这些化合物中,特别优选硫酸锆、硝酸锆等。添加的锆化合物的量优选为0.1-5质量%,更优选为0.2-4质量%,以基于上述超稳定Y-型沸石的氧化锆计。以少于0.1质量%的量添加锆化合物无法改进沸石的固体酸。以超过5质量%的量添加锆化合物可能引起沸石的孔堵塞。通常,通过将锆化合物溶解于水中来制备的锆化合物水溶液适合用作所述锆化合物。上述铪化合物的实例包括氯化铪、硝酸铪、氟化铪、溴化铪、草酸铪等。这些化合物中,特别优选氯化铪、硝酸铪等。添加的铪化合物的量优选为0.1-5质量%,更优选为0.2-4质量%,以基于上述超稳定Y-型沸石的氧化铪计。以少于0.1质量%的量添加铪化合物不能改进沸石的固体酸。以超过4质量%的量添加铪化合物使产生的催化剂昂贵。通常,通过将铪化合物溶解于水中来制备的铪化合物水溶液适合用作所述铪化合物。在此,可以将钛化合物添加上述混合溶液中。钛化合物的实例包括硫酸钛、醋酸钛、氯化钛、硝酸钛、乳酸钛。这些化合物中,特别优选硫酸钛、醋酸钛等。添加的钛化合物的量优选为0.1-5质量%,更优选为0.2-4质量%,以基于上述超稳定Y-型沸石的氧化物计。以少于0.1质量%的量添加钛化合物的添加量导致沸石缺少固体酸位点。以超过5质量%的量添加钛化合物导致沸石的孔堵塞。通常,通过将钛化合物溶解于水中来制备的钛化合物水溶液适合用作所述钛化合物。必须将上述悬浮体的pH预先控制在1.0-2.0,以防止在将锆化合物、铪化合物或钛化合物的水溶液与上述超稳定Y-型沸石的悬浮体混合中产生沉淀。在将锆化合物、铪化合物或钛化合物的水溶液与超稳定Y-型沸石的悬浮体混合的情况下,优选将上述水溶液逐渐添加到悬浮体中。在结束将上述水溶液添加到悬浮体中之后,优选通过搅拌在例如室温(约25℃-约35℃)混合3-5小时。此外,在结束上述混合之后,通过添加碱,诸如氨水等来中和上述混合溶液,以将其pH控制在7.0-7.5,由此可以获得本发明的骨架取代沸石-1。在这方面,当仅将锆化合物(或其水溶液)用作添加到上述悬浮体的化合物(或其水溶液)时,形成了其中形成超稳定Y-型沸石骨架的铝原子的一部分被锆原子取代的骨架取代沸石-1(Zr-USY);当仅使用铪化合物(或其水溶液)时,形成了其中形成超稳定Y-型沸石骨架的铝原子的一部分被铪原子取代的骨架取代沸石-1(Hf-USY);当使用锆化合物和铪化合物(或其水溶液)时,就形成了其中形成超稳定Y-型沸石骨架的铝原子的一部分被锆原子和铪原子取代的骨架取代沸石-1(Zr·Hf-USY)。当将钛化合物(或其水溶液)与添加的锆化合物和/或铪化合物(或它们的水溶液)的组合添加到上述悬浮体中时,形成了其中锆原子、铪原子和钛原子形成超稳定Y-型沸石骨架的一部分的骨架取代沸石-1(Zr·Hf·Ti-USY)。优选将产生的骨架取代沸石-1过滤,如果需要的话,用水洗涤,并在约80℃-约180℃干燥。(5)载体在根据本发明的加氢裂化催化剂中,上述载体包含上述骨架取代沸石-1。上述载体可以包含除上述骨架取代沸石-1之外和/或除上述骨架取代沸石-1之外的骨架取代沸石-2以外的无机氧化物。上述无机氧化物典型地包含用作造粒剂或粘合剂的物质。通常,可以使用包含在包括超稳定Y-型沸石的载体中以及用作造粒剂等的已知物质。作为无机氧化物,可以使用用于相关领域的加氢裂化催化剂和用在加氢处理催化剂中的多孔无机氧化物。其实例包括氧化铝、二氧化硅、氧化钛、二氧化硅-氧化铝、氧化铝-氧化钛、氧化铝-氧化锆、氧化铝-氧化硼、磷-氧化铝、二氧化硅-氧化铝-氧化硼、磷-氧化铝-氧化硼、磷-氧化铝-二氧化硅、二氧化硅-氧化铝-氧化钛和二氧化硅-氧化铝-氧化锆。特别在本发明中,优选主要由氧化铝、二氧化硅-氧化铝组成的无机氧化物。可以根据目的合适地确定载体的骨架取代沸石-1的含量和无机氧化物的含量。载体的骨架取代沸石-1的含量为2-80质量%,优选为20-70质量%,无机氧化物的含量为98-20质量%,优选为80-30质量%。当骨架取代沸石-1和骨架取代沸石-2结合使用时,优选骨架取代沸石-1和骨架取代沸石-2之和的份额少于50%。(6)加氢金属组分作为加氢金属组分,可以使用用于常规加氢裂化催化剂的已知金属组分。其实例包括长周期表第8族的金属组分(铁、钴、镍、铑、钯、银、铱、铂或金)和/或第6族的金属组分(铬、钼或钨)。金属组分的优选实例包括第6族的钼或钨与第8族的钴或镍的组合;和铂族的金属组分(铂、铑、钯等)。加氢裂化催化剂中包含加氢金属组分的量为用于相关领域的加氢裂化催化剂中通常使用的量(以氧化物计0.01-40质量%)。在钼、钨、钴或镍的情况下,它们的量以基于催化剂质量的氧化物计特别优选为3-30质量%。在铂族(铂、铑、钯等)的情况下,它们的量以金属计特别优选为0.01-2质量%。(7)用于烃油的加氢裂化催化剂的性质:根据本发明的加氢裂化催化剂的比表面积优选为200-450m2/g。此外,更适合地建议为250-400m2/g。如果上述比表面积低于200m2/g,则分解率会降低,中间馏分的产率也降低。如果上述比表面积超过450m2/g,则分解率升高,气体馏分趋向增多。在根据本发明的加氢裂化催化剂中,孔径为或以下的孔的体积优选为0.40-0.75ml/g。此外,更适合地建议为0.45-0.70ml/g。如果上述孔体积低于0.40ml/g,则比表面积降低。因此,分解率降低,中间馏分的产率也降低。如果上述孔体积超过0.75ml/g,比表面积增大。因此,分解率升高,气体馏分趋向增多。就此而论,从通过BJH法计算和分析氮气解吸数据获得的孔分布来确定孔体积。在根据本发明的加氢裂化催化剂中,如上所述,优选在其上负载0.01-40质量%的加氢-活性金属组分。在根据本发明的加氢裂化催化剂中包含的锆或铪的量各自分别优选为0.1-5质量%(以氧化物计)。此外,合适地建议为0.5-4质量%。在根据本发明的加氢裂化催化剂中任选包含的钛的量优选为0.1-5质量%(以氧化物计)。此外,合适地建议为0.5-4质量%。用于生产用于烃油的加氢裂化催化剂的方法:根据本发明的用于烃油的加氢裂化催化剂可如下生产:例如,将骨架取代沸石-1与上述无机氧化物(或其前体)混合。通过常规方法将混合物成型为具有期望形状的制品。干燥和烧制该制品以形成载体。通过常规方法用加氢金属组分浸渍载体,干燥和烧制,由此提供加氢裂化催化剂。可选地,将骨架取代沸石-1和无机氧化物(或其前体)与加氢金属组分混合。将混合物成型为期望的形状的制品。干燥和烧制该制品,由此提供加氢裂化催化剂。上述无机氧化物的前体外观为通过与其它催化剂构成成分混合和经过预先处理构成加氢裂化催化剂载体的待成型为无机氧化物的物质。将用于相关领域的此类催化剂的烧制条件应用于烧制载体和加氢裂化催化剂。烧制温度优选为400℃-650℃。通常,根据本发明的氢化催化剂可以通过用含氢化-活性金属组分的水溶液浸渍上述载体,和在400-650℃、在空气中煅烧例如10分钟-3小时来制备。用于加氢裂化烃油的方法:将根据本发明的用于烃油的加氢裂化催化剂装入加氢处理装置(连续反应器)的反应器容器和适合地用于加氢裂化烃油。上述烃油优选包含得自(1)原油、(2)合成原油、(3)沥青、(4)油砂、(5)页岩油或(6)煤馏油的的精制油。适合地用作上述精制油的是选自例如a)真空瓦斯油(VGO),b)得自溶剂脱沥青过程的脱沥青油(DAO)或脱金属油,c)得自焦化过程的轻质焦化瓦斯油或重质焦化瓦斯油,d)得自流化床催化裂化(FCC)过程的循环油或者e)得自减黏裂化过程的瓦斯油的油。加氢裂化可在公知的条件下进行。例如,用上述加氢裂化催化剂填充作为连续反应器反应器装置的加氢处理装置,可以在氢气存在下处理沸点为375℃-833℃的烃油,处理条件为:反应温度为300℃-500℃,氢气压力为4-30MPa,液体时空速(LHSV)为0.1-10h-1和氢气/油比为500-2500Nm3/m3。另外,用上述加氢裂化催化剂填充作为连续反应器的加氢处理,以及可以在氢气存在下处理沸点为375℃-650℃的烃油以获得煤油和瓦斯油,处理条件为:反应器温度为330℃-450℃,氢气压力为7-15MPa,液体时空速(LHSV)为0.2-1.5h-1和氢气/油比为1000-2000Nm3/m3。能够适合地用作上述连续反应器的是选自搅拌浴型反应器、沸腾板型反应器、装有折流板的淤浆床型反应器、固定床型反应器、旋转管型反应器和淤浆床型反应器的连续反应器。根据本发明的用于烃油的加氢裂化催化剂可以特别适合地用于含高沸点馏分的烃的加氢裂化。含高沸点馏分的烃是指其中沸点为560℃或以上的馏分的量占30质量%或以上的烃。含高沸点馏分的烃包括例如真空瓦斯油(VGO)、溶剂脱沥青油(DAO)等。在使用根据本发明的用于烃油的加氢裂化催化剂来加氢裂化烃油,例如含高沸点馏分的烃的情况下,如上所述,由于抑制了煤油-瓦斯油的过度分解反应器,能够以高收率提供中间馏分。用于加氢处理烃油的装置:本发明的用于加氢处理烃油的装置并没有特别限制,只要前述烃油的加氢裂化能够进行即可。可使用多种类型的装置。特别适合的是包括串联的第一催化剂填充罐、第二催化剂填充罐和第三催化剂填充罐的用于加氢处理烃油的装置。每个催化剂填充罐均填充有用于烃油的加氢裂化催化剂。第二催化剂填充罐填充有根据本发明的加氢裂化催化剂。第二催化剂填充罐中的加氢裂化催化剂以相对于填充到第一、第二和第三催化剂填充罐中的全部加氢裂化催化剂的总体积为例如10-60体积%的填充因子使用。然而,填充因子可以不在上述范围内。实施例本发明中使用的分析方法将如下描述。1)组成分析:使用X射线荧光分析仪(RigakuCorporation制造的“RIX3000”)进行样品(沸石或加氢裂化催化剂)的组成分析(Zr、Hf、Ti、Mo或Ni)。通过玻璃珠法制备用于测量的样品。具体而言,将5g样品放入环内径为35mm的氯乙烯制的环中,借助压力成型机施加20t的压力模压20秒以制备用于测量的样品。X射线荧光分析的条件如下:靶:Rh,分析晶体:LiF,检测器:闪烁计数器,激发:4kW的Rh管,测量电压:55kV,电流:70mA。2)沸石中的钠的检测:使用原子吸收光谱仪(HORIBALtd.制造的“Z5300”)测量样品(沸石)中的钠含量。将测量波长范围控制在190-900nm。3)晶格常数:使用X射线衍射仪(RigakuCorporation制造的“RINT2100”)测量样品(沸石)的X射线衍射峰,由它的结果计算晶格常数。计算晶格常数的方法已描述在本说明书中。X射线衍射的条件如下:导管:Cu-K(α线),2θ扫描范围:20-50°,扫描速度:0.01°/分钟,扫描步长:0.01°。4)结晶度:由样品(沸石)的X射线衍射峰计算结晶度。它的计算方法已描述在本说明书中。5)SiO2/Al2O3摩尔比:由样品(沸石)的X射线衍射峰确定Si和Al的峰强度比,将它折算为SiO2/Al2O3的摩尔比。6)比表面积和孔体积:使用吸附测量设备(由QuantachromeInstrumentsCorporate制造的全自动气体吸附设备“AUTOSORB-1”)对0.02-0.05g样品(沸石或加氢裂化催化剂)在室温下脱气处理5小时,然后在液氮温度下测量吸附解吸等温线以使用多点法BET方程计算单位质量的比表面积。另外,由通过BJH法的氮气吸附等温线计算孔分布和孔体积(孔径:或以下)。7)紫外线-可见光/近红外线/分光光度法(UV-vis-NIR光谱):借助紫外线-可见光/近红外线/分光光度仪(型号:JASCOV-570,JASCOCorporation制造)测量沸石的UV-vis-NIR光谱。通过将溴化钾与样品以99:1的比例进行物理混合,并在500kg/cm2的压力下将50mg该混合物模压为薄片形式制备样品。然后以3.0℃/分钟的加热速率将经模压的物质加热至200℃,和通过抽真空3小时进行预处理,然后在室温下进行测量,测量条件为:光谱带宽为10mm和扫描速度为400nm/分钟。8)傅里叶变换红外光谱法(FT-IR光谱):借助透射傅里叶变换红外分光镜(JIR-7000,JASCOCorporation制造)测量沸石的羟基基团的FT-IR光谱和骨架振动面积。通过在500kg/m2的压力下将20mg样品模压为薄片形式来制备样品。其后以6.7℃/分钟的加热速率将经模压的物质加热至400℃,和通过抽真空3小时进行预处理,然后在室温下进行测量,测量条件为:分辨率为4cm-1和积分频率为500次。表格说明:表1:表1中显示了用作原料的USY(a)-(m)的性质。表2:表2中显示了用于实施例1-实施例6的骨架取代沸石-1(USY(A)-USY(F))的性质。表3:表3中显示了实施例1-实施例6中制备的加氢裂化催化剂(催化剂A-催化剂F)的性质。表4:表4中显示了测试实施例中使用的原料油的性质。表5:表5中显示了根据本发明的加氢裂化催化剂的测试结果(相对裂化率和相对中间馏分收率)。实施例1:加氢裂化催化剂A超稳定Y沸石首先,将50.0kg的SiO2/Al2O3摩尔比为5.2、晶胞大小(UD)为2.466nm、比表面积(SA)为720m2/g和Na2O含量为13.0质量%的NaY沸石(下文也称为“NaY”)悬浮于500升(以下也表示为“L”)温度为60℃的水中。此外,向其中添加14.0kg硫酸铵。将得到的悬浮体在70℃搅拌1小时并过滤。用水洗涤产生的固体。然后用将14.0kg硫酸铵溶解于500L温度为60℃的水中的硫酸铵溶液洗涤固体,用500L温度为60℃的水洗涤,在130℃干燥20小时,由此提供约45kg的Y沸石(NH465Y),其中NaY中包含的65%的钠(Na)被铵离子(NH4+)离子交换。NH465Y中Na2O的含量为4.5质量%。在饱和水蒸气压气氛中于670℃将40kg的NH465Y烧制1小时以形成氢-Y沸石(HY)。将HY悬浮于400L温度为60℃的水中。然后向其中添加49.0kg的硫酸铵。在90℃将产生的混合物搅拌1小时,用200L温度为60℃的水洗涤。然后在130℃将混合物干燥20小时,由此提供约37kg的Y沸石(NH495Y),其中初始的NaY中包含的95%的Na被NH4离子交换。在饱和水蒸气压气氛中于650℃将33.0kg的NH495Y烧制1小时,由此提供约15kg的SiO2/Al2O3摩尔比为5.2和Na2O含量为0.60质量%的超稳定Y沸石(下文也称为“USY(a)”)。表1显示了USY(a)的物理性质。接着,将26.0kg的该USY(a)悬浮于260L温度为60℃的水中。在逐渐将61.0kg的25质量%硫酸添加到悬浮体中后,在70℃将该悬浮体搅拌1小时。过滤该悬浮体。用260升温度为60℃的去离子水洗涤产生的固体,并在130℃干燥20小时,由此提供超稳定Y-型沸石(下文也称为“USY(b)”)。表1显示了USY(b)的物理性质。在600℃下将USY(b)烧制1小时,由此提供约17kg的超稳定Y-型沸石(下文也称为“USY(c)”)。表1显示了USY(c)的物理性质。锆-取代沸石:USY(A)的制备首先,将1kg的USY(c)悬浮于10L温度为25℃的水中。用25质量%的硫酸将悬浮体的pH调至1.6。然后向其中添加86g含18质量%硫酸锆的溶液。在室温下将产生的混合物搅拌3小时。然后通过15质量%的氨水将pH调至7.2。在室温下将混合物搅拌1小时后,过滤混合物。用10L水洗涤产生的固体,并在130℃下干燥20小时,由此提供约1kg的锆-取代沸石(下文也称为“USY(A)”)。表2显示了USY(A)的物理性质。图1显示了USY(A)的UV光谱。图1证明了如下内容:来源于用作USY(A)原料的硫酸锆的ZrO2的UV光谱在约230和约280nm显示峰,而USY(A)的UV光谱在约200-220nm显示峰。因此,USY(A)的骨架被Zr取代。在此,用紫外线-可见光-近红外线分光光度仪(JASCOV-570,JASCOCorporation制造)在10mm的带宽和400nm/min的扫描速率下测量沸石的UV光谱。同时,将50mg溴化钠与USY(A)之比为99:1的混合物在500kg/cm2的压力下形成为水样UV-光谱样品。将该样品置于紫外线-可见光-近红外线分光光度仪中。在通过在3℃/min的加热速率下将样品加热至200℃和将样品抽空3小时进行的预处理后,在室温下以4cm-1的分辨率和500的积分数下进行测量。使用X射线荧光分析仪(RIX3000,RigakuCorporation制造)进行沸石的组成分析。通过玻璃珠法制备样品。采用原子吸收光谱仪(Z-5300,HORIBA,Ltd制造)测量沸石中的钠。采用X射线衍射仪(RINT2100,RigakuCorporation制造)测量结晶度和晶格常数。采用孔分布分析仪(Autosorb,QuantachromeInstruments制造)测量比表面积和孔体积。加氢裂化催化剂A首先,将40kg的以Al2O3计6.8质量%铝酸钠的水溶液与40kg的以Al2O3计2.4质量%硫酸铝的水溶液混合。再在60℃下将混合物搅拌1小时,然后用150L的0.3质量%氨水溶液洗涤产物以除去Na2SO4。接着,将水添加除去了Na2SO4的产物以将Al2O3浓度调至10质量%。通过15质量%的氨水将pH调至10.5。在95℃将混合物搅拌10小时,脱水,洗涤和用捏合机捏合,由此提供氧化铝混合物。将产生的氧化铝混合物与USY(A)以1:1的干质量比混合。用捏合机捏合混合物,成型为直径为1.8mm的圆柱形,在550℃下烧制3小时,由此提供载体A。将载体A浸入含氢化-活性金属组分的水溶液中,并在550℃下、空气中烧制1小时,由此提供加氢裂化催化剂A。在此,含氢化-活性金属组分的水溶液如下制备:将700mL水添加到201.3g三氧化钼(氢化-活性金属组分的实例)和90.4g碳酸镍(氢化-活性金属组分的实例)中,并在95℃下将产生的混合物搅拌5小时。加氢裂化催化剂A以氧化物计包含0.39质量%的锆、16.7质量%的钼和3.88质量%的镍。表3显示了加氢裂化催化剂A的物理性质。实施例2:加氢裂化催化剂B将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加8g氯化铪并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.0-7.5,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg铪-取代型沸石(以下称为“USY(B)”)。它的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(B)的加氢裂化催化剂B。加氢裂化催化剂B的性质示于表3中。实施例3:加氢裂化催化剂C将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.5-1.7。添加86g18质量%的硫酸锆和8g氯化铪并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆·铪-取代型沸石(以下称为“USY(C)”)。它的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(C)的加氢裂化催化剂C。加氢裂化催化剂C的性质示于表3中。实施例4:加氢裂化催化剂D将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆、8g氯化铪和60g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆·铪·钛-取代型沸石(以下称为“USY(D)”)。USY(D)的性质示于表2中,它的UV光谱示于图2中。如图2所示,确认了得自作为原料的硫酸氧钛的TiO2的UV光谱在220和320nm附近具有峰和USY(D)由于Zr和Ti的取代在210-320nm附近具有峰。另外,以如实施例1中相同的方式制备含USY(D)的加氢裂化催化剂D。加氢裂化催化剂D的性质示于表3中。实施例5:加氢裂化催化剂E将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆和60g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆·钛-取代型沸石(以下称为“USY(E)”)。USY(E)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(E)的加氢裂化催化剂E。加氢裂化催化剂E的性质示于表3中。实施例6:加氢裂化催化剂F将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加8g氯化铪和60g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg铪·钛-取代型沸石(以下称为“USY(F)”)。USY(F)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(F)的加氢裂化催化剂F。加氢裂化催化剂F的性质示于表3中。比较例1:加氢裂化催化剂G将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加60g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg钛-取代型沸石(以下称为“USY(G)”)。USY(G)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(G)的加氢裂化催化剂G。加氢裂化催化剂G的性质示于表3中。比较例2:加氢裂化催化剂H将1kg在实施例1中获得的煅烧前的USY(b)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(H)”)。USY(H)的性质示于表2中,USY(H)的UV光谱示于图3。在图3中,从USY(H)和USY(A)的UV光谱的比较可以观察到,通过煅烧加速了锆的取代。另外,以如实施例1中相同的方式制备含USY(H)的加氢裂化催化剂H。加氢裂化催化剂H的性质示于表3中。比较例3:加氢裂化催化剂I将1kg通过将在实施例1中获得的USY(b)在400℃下煅烧1小时制备的超稳定Y-型沸石(以下称为“USY(d)”)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(I)”)。在这方面,USY(d)的性质示于表1;USY(I)的性质示于表2中;USY(I)的UV光谱示于图3中。在图3中,从USY(I)和USY(A)的UV光谱的比较发现,通过在400℃下的煅烧未加速锆的取代。另外,以如实施例1中相同的方式制备含USY(I)的加氢裂化催化剂I。加氢裂化催化剂I的性质示于表3中。比较例4:加氢裂化催化剂J将1kg通过将在实施例1中获得的USY(b)在800℃下煅烧1小时制备的超稳定Y-型沸石(以下称为“USY(e)”)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(J)”)。USY(e)的性质示于表1中;USY(J)的性质示于表2中;USY(J)的UV光谱示于图3中。另外,以如实施例1中相同的方式制备含USY(J)的加氢裂化催化剂J。加氢裂化催化剂J的性质示于表3中。比较例5:加氢裂化催化剂K将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至0.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(K)”)。USY(K)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(K)的加氢裂化催化剂K。加氢裂化催化剂K的性质示于表3中。比较例6:加氢裂化催化剂L将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至2.4。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(L)”)。USY(L)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(L)的加氢裂化催化剂L。加氢裂化催化剂L的性质示于表3中。比较例7:加氢裂化催化剂M将2kg在实施例1中获得的USY(a)悬浮于20L60℃的温水中。将3.7kg25质量%的硫酸逐渐添加到上述悬浮体中,然后在70℃下搅拌1小时以溶解骨架外的铝。然后,过滤悬浮体,用20升60℃的净化水洗涤获得的物质,在130℃下干燥20小时以获得超稳定Y-型沸石(以下称为“USY(f)”)。USY(f)的性质示于表1中。将由此获得的USY(f)在600℃下煅烧1小时以获得约1.5kg超稳定Y-型沸石(以下称为“USY(g)”)。USY(g)的性质示于表1中。将1kg由此得到的USY(g)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(M)”)。USY(M)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(M)的加氢裂化催化剂M。加氢裂化催化剂M的性质示于表3中。比较例8:加氢裂化催化剂N将2kg在实施例1中获得的USY(a)悬浮于20L60℃的温水中。将13.6kg25质量%的硫酸逐渐添加到上述悬浮体中,然后在70℃下搅拌1小时以溶解骨架外的铝。然后,过滤悬浮体,用20升60℃的净化水洗涤获得的物质,在130℃下干燥20小时以得到超稳定Y-型沸石(以下称为“USY(h)”)。USY(h)的性质示于表1中。将由此获得的USY(h)在600℃下煅烧1小时以获得约11kg超稳定Y-型沸石(以下称为“USY(i)”)。USY(i)的性质示于表1中。将1kg由此获得的USY(i)悬浮于10L25℃的水中,用25质量%的硫酸将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(N)”)。USY(N)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(N)的加氢裂化催化剂N。加氢裂化催化剂N的性质示于表3中。比较例9:加氢裂化催化剂O将2kg在实施例1中获得的NH495Y在800℃下、饱和水蒸气气氛中煅烧1小时以获得约15kgUD为2.425nm的超稳定Y-型沸石(以下称为“USY(j)”)。USY(j)的性质示于表1中。将1kg由此获得的USY(j)在600℃下煅烧1小时以得到1.5kg超稳定Y-型沸石(以下称为“USY(k)”)。USY(k)的性质示于表1中。将1kg由此获得的USY(k)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(O)”)。USY(O)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(O)的加氢裂化催化剂O。加氢裂化催化剂O的性质示于表3中。比较例10:加氢裂化催化剂P将2kg在实施例1中得到的NH495Y在800℃下、饱和水蒸气气氛中煅烧1小时以得到UD为2.455nm的超稳定Y-型沸石(以下称为“USY(l)”)。USY(l)的性质示于表1中。将由此获得的USY(l)在600℃下煅烧1小时以获得约1.5kg超稳定Y-型沸石(以下称为“USY(m)”)。USY(m)的性质示于表1中。将1kg由此获得的USY(m)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加86g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(P)”)。USY(P)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(P)的加氢裂化催化剂P。加氢裂化催化剂P的性质示于表3中。比较例11:加氢裂化催化剂Q将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加8.6g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(Q)”)。USY(Q)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(Q)的加氢裂化催化剂Q。加氢裂化催化剂Q的性质示于表3中。比较例12:加氢裂化催化剂R将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加516g18质量%的硫酸锆并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆-取代型沸石(以下称为“USY(Q)”)。USY(Q)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(Q)的加氢裂化催化剂Q。加氢裂化催化剂Q的性质示于表3中。比较例13:加氢裂化催化剂S将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加1.6g氯化铪并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg铪-取代型沸石(以下称为“USY(S)”)。USY(S)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(S)的加氢裂化催化剂S。加氢裂化催化剂S的性质示于表3中。比较例14:加氢裂化催化剂T将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加96g氯化铪并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg铪-取代型沸石(以下称为“USY(T)”)。USY(T)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(T)的加氢裂化催化剂T。加氢裂化催化剂T的性质示于表3中。比较例15:加氢裂化催化剂U将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加86g18质量%的硫酸锆和6.0g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆·钛-取代型沸石(以下称为“USY(U)”)。USY(U)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(U)的加氢裂化催化剂U。加氢裂化催化剂U的性质示于表3中。比较例16:加氢裂化催化剂V将1kg在实施例1中获得的USY(c)悬浮于10L25℃的水中,用25质量%的硫酸水溶液将溶液的pH调至1.6。添加86g18质量%的硫酸锆和516g33质量%的硫酸氧钛并混合,在室温下将悬浮体搅拌3小时。然后,通过15质量%的氨水将pH调至7.2,在室温下将悬浮体搅拌1小时,然后过滤。用10L水洗涤获得的物质,在130℃下干燥20小时以获得约1kg锆·钛-取代型沸石(以下称为“USY(V)”)。USY(V)的性质示于表2中。另外,以如实施例1中相同的方式制备含USY(V)的加氢裂化催化剂V。加氢裂化催化剂V的性质示于表3中。表1USY二氧化硅-氧化铝之比晶格常数(nm)比表面积(m2/g)结晶度(%)a5.22.43863598b30.22.436710105c30.12.436712105d30.22.438710105e30.12.435708103f15.52.437705104g15.62.437702103h1262.434703101i1252.434705101j30.82.42858588k30.52.42858387l29.52.45568598m29.32.45468897表2表3测试例:催化剂活性评价使用催化剂A-X和具有表4中所示性质的烃油作为原料,通过XytelCorporation制造的固定床连续型反应器设备进行加氢裂化反应器,条件为:氢气分压为13MPa,液体时空速为0.26hr-1,氢油比(氢气/油之比)为1250Nm3/kL和反应器温度为370℃,来根据以下方程式(2)确定分解速率,和根据以下方程式(3)确定中间馏分(煤油和轻油)收率。根据如下程序1)和2),基于分解速率和中间馏分收率的值来评价催化剂活性。1)确定其它催化剂的分解速率与催化剂A的分解速率的比并显示为相对分解速率,其中将催化剂A的分解速率设为100。结果示于表5中。2)确定其它催化剂的中间馏分收率与催化剂A的中间馏分收率的比并显示为相对中间馏分收率,其中将催化剂A的中间馏分收率设为100。结果示于表5中。分解速率(%)=(产出油中沸点高于375℃的部分的含量(kg))/(原料油中沸点高于375℃的部分的含量(kg))×100(2)中间馏分的收率(%)=(产出油中沸点为149-375℃的部分的含量(kg))/(原料油中沸点低于375℃的部分的含量(kg))×100(3)在此,分解速率和中间馏分收率的“%”都是指“质量%”。根据表5显示的结果,根据本发明的催化剂(催化剂A-F)的分解速率和中间馏分收率的至少一个高于比较例的催化剂(催化剂G-V),上述数值之外的其它值至少与之相当。这意味着显示了根据本发明的催化剂的优越性。表4比重(g/ml)0.9203硫含量(质量%)2.23氮含量(重量ppm)815C5-85℃(质量%)085-149℃(质量%)0149-185℃(质量%)0185-240℃(质量%)1.3240-315℃(质量%)2.7315-375℃(质量%)8.0375-560℃(质量%)79.5560℃+(质量%)8.5表5催化剂相对分解速率(%)相对中间馏分收率(%)实施例1A100100实施例2B9997实施例3C9996实施例4D10095实施例5E9996实施例6F10096比较例1G9991比较例2H9893比较例3I9792比较例4J9689比较例5K9891比较例6L9891比较例7M9791比较例8N9989比较例9O9990比较例10P9792比较例11Q9994比较例12R9893比较例13S9894比较例14T9792比较例15U9894比较例16V9590实施例7:加氢裂化催化剂W通过将具有8.5质量%二氧化硅的水玻璃添加到25质量%的硫酸水溶液中,获得具有7质量%二氧化硅的硅胶。另一方面,通过将40kg具有6.8质量%Al2O3的二氧化铝钠水溶液与40kg具有2.4质量%Al2O3的硫酸铝水溶液混合,获得氧化铝浆料。将上述硅胶与氧化铝浆料以70:30的质量比混合,并在60℃下搅拌1小时。过滤后,用150ml的0.3质量%氨的铵水溶液洗涤产物以除去Na2SO4。再通过水稀释产物以生产10质量%水的浆料,并添加15质量%的铵水溶液将它的pH控制在10.5。然后,在95℃下搅拌10小时,除去水,洗涤和捏合以提供二氧化硅-氧化铝产物。将获得的二氧化硅-氧化铝与USY(A)以二氧化硅-氧化铝:USY=1.5:1的比例(以干质量计)进行混合,然后捏合,挤出为1.8mm直径的圆柱形,干燥,在550℃下煅烧3小时以提供载体W。另外,以如实施例1中相同的方式制备加氢裂化催化剂W。加氢裂化催化剂W的组成和物理性质示于表6中。表6实施例8:加氢裂化催化剂X通过将具有8.5质量%二氧化硅的水玻璃添加到25质量%的硫酸水溶液中,获得具有7质量%二氧化硅的硅胶。另一方面,将40kg具有6.8质量%Al2O3的二氧化铝钠水溶液与40kg具有2.4质量%Al2O3的硫酸铝水溶液混合,得到氧化铝浆料。将上述硅胶与氧化铝浆料以70:30的质量比混合,并在60℃下搅拌1小时。过滤后,用150ml的0.3质量%氨的铵水溶液洗涤产物以除去Na2SO4。再用水稀释产物以生产含10质量%水的浆料,并添加15质量%的铵水溶液将它的pH控制在10.5。然后,在95℃下搅拌10小时,除去水,洗涤和捏合以提供二氧化硅-氧化铝产物。将得到的二氧化硅-氧化铝与USY(A)以二氧化硅-氧化铝:USY=2:1的比例(以干质量计)进行混合,然后捏合,挤出为1.8mm直径的圆柱形,干燥,在550℃下煅烧3小时以提供载体X。另外,以如实施例1中相同的方式制备加氢裂化催化剂X。加氢裂化催化剂X的组成和物理性质示于表6中。实施例9:加氢裂化催化剂Y通过将具有8.5质量%二氧化硅的水玻璃添加到25质量%的硫酸水溶液中,得到具有7质量%二氧化硅的硅胶。另外,将40kg具有6.8质量%Al2O3的二氧化铝钠水溶液与40kg具有2.4质量%Al2O3的硫酸铝水溶液混合,得到氧化铝浆料。将上述硅胶与氧化铝浆料以30:70的质量比混合,并在60℃下搅拌1小时。过滤后,用150ml的0.3质量%氨的铵水溶液洗涤产物以除去Na2SO4。再用水稀释产物以生产含10质量%水的浆料,并添加15质量%的铵水溶液将它的pH控制在10.5。然后,在95℃下搅拌10小时,除去水,洗涤和捏合以提供二氧化硅-氧化铝产品。将得到的二氧化硅-氧化铝与USY(A)以二氧化硅-氧化铝:USY=2:1的比例(以干质量计)进行混合,然后捏合,挤出为1.8mm直径的圆柱形,干燥,在550℃下煅烧3小时以提供载体Y。另外,以如实施例1中相同的方式制备加氢裂化催化剂Y。加氢裂化催化剂Y的组成和物理性质示于表6中。通过使用与上述相同的方法评价制得的催化剂W、X和Y的活性。结果示于表7。表7催化剂相对分解速率(%)相对中间馏分收率(%)实施例7W10298实施例8X98106实施例9Y102105如在表7中可见,具有氧化铝-二氧化硅作为载体的催化剂同具有氧化铝载体的一样取得了高裂化活性和中间馏分选择性。一般而言,与氧化铝相比,由于氧化铝-二氧化硅具有相对更强的酸性位点,尽管USY沸石含量较低,但裂化活性更高。结果是,氧化铝-二氧化硅比氧化铝与USY沸石的中间馏分选择性更高。本发明不应仅限于上述实施方案,只要本发明的范围不变,可以对本发明进行变换。例如,将各上述实施方案和各变形实施例的部分或全部进行组合从而构成根据本发明的用于烃油的加氢裂化催化剂,和使用了上述催化剂的加氢处理方法,也都包括在本发明的权利范围之内。例如,在上述实施方案中用于烃油的加氢裂化催化剂中,根据本发明的用于烃油的加氢裂化催化剂可预先与催化剂(Y)混合,从而用混合催化剂填充各阶段的催化剂填充浴。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种铼聚合离子液体催化烯烃环氧化的方法与制造工艺
- 一种制备高纯度烯烃的催化剂的制备方法与制造工艺
- 稀土催化剂组合物及其制备方法和共轭二烯烃的聚合方法与制造工艺
- 一种催化剂的制备方法和制得的催化剂及其应用与制造工艺
- 一种用于烯烃聚合的催化剂体系及其应用的制造方法与工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分、催化剂及其应用的制造方法与工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺