一种氧化石墨烯-共价有机骨架材料复合材料、毛细管电色谱柱及制备方法与流程
本发明属于化学合成及分析技术领域,特别涉及一种氧化石墨烯-共价有机骨架材料复合材料、毛细管电色谱柱及制备方法。
背景技术:
磺胺类药物是一类人工合成的抗菌类药物,由于其抗菌谱广且价格低廉,在药物领域有广泛应用,特别是作为兽药。但如果过量使用,会导致药物残留在动物体内,随着食物链进入到人体当中,对人体产生危害,甚至可能会导致细菌的耐药性。因此,建立环境水样以及动物源性食品中磺胺类药物的分析方法尤为重要。目前常用于分离磺胺类药物的方法为毛细管电泳法,即毛细管区带电泳,该方法通常使用内径为50-100μm的未修饰的石英毛细管柱分离磺胺,目前的毛细管电泳法分离磺胺类药物存在分离度低、分离效率低等问题。
目前共价有机骨架材料的制备过程一般采用溶剂热法合成共价有机骨架材料,该方法需要在较高温度下封闭条件内进行,反应条件苛刻,并且反应时间较长(密闭容器高温加热2天以上),制备工艺复杂,且共价有机骨架材料的性能一致性较差。
技术实现要素:
鉴于以上分析,本发明旨在提供一种氧化石墨烯-共价有机骨架材料复合材料、毛细管电色谱柱及制备方法。至少能够解决以下技术问题之一:(1)现有的毛细管电泳法分离磺胺类药物分离度低;(2)分离效率低。
本发明的目的主要是通过以下技术方案实现的:
一方面,本发明提供了一种氧化石墨烯-共价有机骨架材料复合材料,氧化石墨烯-共价有机骨架复合材料的制备原料包括氧化石墨烯、反应溶剂和共价有机骨架材料单体。
进一步的,氧化石墨烯、反应溶剂和共价有机骨架材料单体的质量体积比为:30-40mg:40-50ml:50-60mg。
进一步的,共价有机骨架材料单体的制备原料包括三醛基间苯三酚和联苯二胺;三醛基间苯三酚和联苯二胺的质量比为:20-35:25-30。
本发明还提供了一种氧化石墨烯-共价有机骨架材料复合材料的制备方法,包括如下步骤:
步骤1:将氧化石墨烯在反应溶剂中超声分散均匀;
步骤2:加入三醛基间苯三酚,超声完全溶解,得到第一混合悬浊液;
步骤3:将第一混合悬浊液离心,去除上清液,再加入联苯二胺和反应溶剂进行超声反应得到第二混合悬浊液;
步骤4:将第二混合悬浊液离心,去除上清液,再加入三醛基间苯三酚和联苯二胺,加入反应溶剂进行超声反应,得到含有氧化石墨烯-共价有机骨架材料的第三混合悬浊液;
步骤5:将第三混合悬浊液冷却到室温,离心,去除上清液,清洗下层的氧化石墨烯-共价有机骨架材料,然后烘干,得到氧化石墨烯-共价有机骨架材料复合材料固体。
进一步的,步骤5中,烘干温度为60-70℃,烘干时间为6-8h。
本发明还提供了一种毛细管电色谱柱,毛细管电色谱柱包括氧化石墨烯-共价有机骨架材料复合材料。
本发明还提供了一种毛细管电色谱柱的制备方法,包括以下步骤:
步骤a:将氧化石墨烯-共价有机骨架材料复合材料溶于反应试剂中,超声分散,得到第一混合液;
步骤b:在第一混合液中加入脱水剂和3-氨基丙基三乙氧基硅烷,超声充分溶解,静置后离心去除上层清液,得到黑色固体;
步骤c:黑色固体使用反应试剂清洗,烘干,得到3-氨基丙基三乙氧基硅烷修饰的氧化石墨烯-共价有机骨架材料复合材料;
步骤d:将石英毛细管柱活化,氮气吹干;
步骤e:将3-氨基丙基三乙氧基硅烷修饰氧化石墨烯-共价有机骨架材料复合材料分散在分散溶剂中,得到悬浊液,将悬浊液注入毛细管柱内静置,氮气吹干,静置,得到氧化石墨烯-共价有机骨架材料复合材料作为固定相的毛细管电色谱柱。
进一步的,步骤a中,反应试剂为n,n-二甲基甲酰胺。
进一步的,氧化石墨烯-共价有机骨架材料复合材料的质量与反应试剂的体积比为10-15mg:5-10ml。
进一步的,步骤b中,第一混合液、脱水剂和3-氨基丙基三乙氧基硅烷的质量体积比为:5-10ml:50-80mg:0.1-0.2ml。
与现有技术相比,本发明至少能实现以下技术效果之一:
1)本发明首次利用三醛基间苯三酚、联苯二胺、氧化石墨烯,通过超声合成法制备了氧化石墨烯-共价有机骨架材料复合材料,该方法简单方便,避免了现有技术中使用加热等方法进行材料的制备,有利于简便的制备材料;本发明的氧化石墨烯-共价有机骨架材料复合材料的比表面积较大、孔隙率较大、密度较小,使用范围广。
2)本发明采用超声合成法制备氧化石墨烯-共价有机骨架材料复合材料,大大缩短了材料制备的时间(由原来的2天以上减少到现在的7h50min-10h55min),能耗节省60%以上。
3)本发明的氧化石墨烯-共价有机骨架材料复合材料能够用作毛细管电色谱柱固定相,利用3-氨基丙基三乙氧基硅烷作为连接体,通过化学键合法制备的氧化石墨烯-共价有机骨架材料为固定相的毛细管电色谱柱,由于生成的化学键是相对稳定的,相较于动态涂布法及物理吸附法,该方法较为稳定,该方法制备的毛细管电色谱柱稳定性好,通过对分离磺胺类药物的日内及日间精密度进行评价,十种磺胺类药物峰面积的日内相对标准偏差在0.33%-3.64%之间,日间精密度在1.65%-7.16%之间。
4)本发明提供的氧化石墨烯-共价有机骨架材料复合材料为固定相的毛细管电色谱柱在磺胺类药物的毛细管电色谱分析中,提高了分离度、缩短分析时间、提升分离效率,能够应用于其他药物的毛细管电色谱分析检测以及其他色谱领域。
本发明的其他特征和优点将在随后的说明书中阐述,并且,部分可从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在所写的说明书以及附图中所特别指出的结构来实现和获得。
附图说明
附图仅用于示出具体实施例的目的,而并不认为是对本发明的限制,在整个附图中,相同的附图标记表示相同的部件。
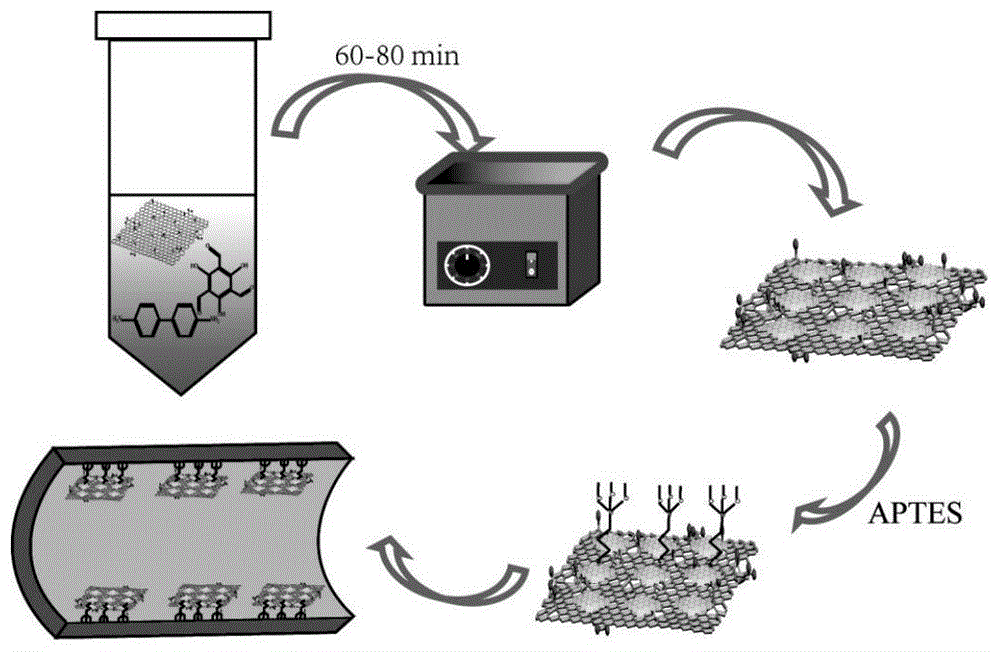
图1为制备本发明的毛细管电色谱柱的工艺流程示意图;
图2为本发明的氧化石墨烯-共价有机骨架材料复合材料的扫描电镜图;
图3为本发明的共价有机骨架材料的扫描电镜图;
图4为本发明的毛细管电色谱柱对十种磺胺类药物进行分析检测的色谱图;
图5为本发明的毛细管电色谱柱的扫描电镜图;
图6为普通的毛细管柱的扫描电镜图。
附图标记:
1-磺胺二甲嘧啶(sdd);2-磺胺索嘧啶(ssd);3-磺胺甲氧哒嗪(smp);4-磺胺甲基嘧啶(smr);5-磺胺甲氧哒嗪(sdz);6-磺胺间甲氧嘧啶(smt);7-磺胺氯哒嗪(scd);8-磺胺索嘧啶(ssd);9-酞磺胺噻唑(pst);10-磺胺噻唑(st)。
具体实施方式
以下结合具体实施例对一种氧化石墨烯-共价有机骨架材料复合材料、毛细管电色谱柱及制备方法作进一步的详细描述,这些实施例只用于比较和解释的目的,本发明不限定于这些实施例中。
需要说明的是,在本发明中诸如第一、第二等之类的关系术语仅仅用来将一个实体或操作与另一个实体或操作区分开来,而不一定暗示或者要求这些实体或操作之间存在任何这种实际的关系或顺序。
本发明提供了一种氧化石墨烯-共价有机骨架材料复合材料,氧化石墨烯-共价有机骨架复合材料的制备原料包括氧化石墨烯、反应溶剂和共价有机骨架材料单体。
具体的,考虑到氧化石墨烯含量过高会导致共价有机骨架材料在氧化石墨烯上分布较少;过低会导致共价有机骨架材料自身发生堆叠。因此,控制氧化石墨烯、反应溶剂和共价有机骨架材料单体的质量体积比为:30-40mg:40-50ml:50-60mg。
具体的,反应溶剂为乙醇和四氢呋喃的混合溶液;其中,乙醇和四氢呋喃的体积比为9:1。
具体的,共价有机骨架材料单体的制备原料包括三醛基间苯三酚和联苯二胺;考虑到三醛基间苯三酚和联苯二胺的质量比过大或过小均会导致剩余的单体无法合成共价有机骨架材料,导致单体的浪费,其中,联苯二胺质量过小会导致没有足量的氨基与氧化石墨烯上的羧基进行反应。因此,控制三醛基间苯三酚和联苯二胺的质量比为:20-35:25-30。
与现有技术相比,本发明中采用三醛基间苯三酚、联苯二胺能够合成tpbd型共价有机骨架材料,共价有机骨架材料中的氨基会和氧化石墨烯中的羧基进行反应,使氧化石墨烯-共价有机骨架材料复合材料合成。本发明的氧化石墨烯-共价有机骨架材料复合材料的比表面积大、孔隙率大、密度低,使用范围广泛。
本发明还提供了一种氧化石墨烯-共价有机骨架材料复合材料的制备方法,包括以下步骤:
步骤1:将氧化石墨烯在反应溶剂中超声分散均匀;
步骤2:加入三醛基间苯三酚,超声充分溶解,得到第一混合悬浊液;
步骤3:将第一混合悬浊液离心,去除上清液,再加入联苯二胺、催化剂和反应溶剂进行超声反应得到第二混合悬浊液;
步骤4:将第二混合悬浊液离心,去除上清液,再加入三醛基间苯三酚、联苯二胺和催化剂,加入反应溶剂进行超声反应,得到含有氧化石墨烯-共价有机骨架材料的第三混合悬浊液;
步骤5:将第三混合悬浊液冷却到室温,离心,去除上清液,清洗下层的氧化石墨烯-共价有机骨架材料,然后烘干,得到氧化石墨烯-共价有机骨架材料复合材料固体。
具体的,上述步骤1和步骤3中的反应溶剂均为乙醇和四氢呋喃的混合溶液;其中,乙醇和四氢呋喃的体积比为9:1。
具体的,步骤3和步骤4中为了促进三醛基间苯三酚与联苯二胺充分反应,加入催化剂,具体的,催化剂为三氟乙酸。
具体的,上述步骤2-4中,为了保证tpbd型共价有机骨架材料尽可能均匀生长在氧化石墨烯表面,要先加入一部分三醛基间苯三酚(即第一次三醛基间苯三酚),再加入一部分联苯二胺(即第一次联苯二胺)和反应溶剂,最后再加入一部分三醛基间苯三酚(即第二次三醛基间苯三酚)和联苯二胺(即第二次联苯二胺)。
具体的,上述步骤2-4中,第一次三醛基间苯三酚、第一次联苯二胺、第二次三醛基间苯三酚及第二次联苯二胺的质量比为:20-35:25-30:20-35:25-30。
具体的,上述步骤3中,联苯二胺(即第一次联苯二胺)与催化剂的质量体积比为25-30mg:0.15-0.25ml。
具体的,上述步骤4中,三醛基间苯三酚(即第二次三醛基间苯三酚)、联苯二胺(即第二次联苯二胺)和催化剂的质量体积比为20-35mg:25-30mg:0.15-0.25ml。
具体的,上述步骤1中,超声分散时间过长会影响材料性质;过短会使氧化石墨烯在溶剂中分散不均匀。因此,控制超声分散时间为20-40min。
具体的,步骤2中,超声时间过长会影响材料性质;超声时间过短会导致三醛基间苯三酚与氧化石墨烯结合不完全。因此,控制超声时间为15-20min。
具体的,步骤3中,离心转速过大会浪费能源,并使材料不易在下一步分散;过小会导致离心不完全;离心时间过长会导致材料不易在下一步分散,离心时间过短会导致离心不完全。因此,控制离心转速为7000-9000rpm,离心时间为5-10min,超声时间为5-15min。
具体的,步骤4中,离心转速过大会浪费能源,并使材料不易在下一步分散;过小会导致离心不完全;离心时间过长会导致材料不易在下一步分散,离心时间过短会导致离心不完全。因此,控制离心转速为7000-9000rpm,离心时间为5-10min,超声时间为60-80min。
具体的,上述步骤5中,离心转速为7000-9000rpm,离心时间为5-10min,清洗后烘干包括如下步骤:用四氢呋喃和乙醇各洗3遍,60-70℃烘干6-8h。其中,烘干温度过高会导致材料的分解,过低会使材料无法烘干;烘干时间过长使材料晶型发生改变;过短会使材料烘干不完全。因此,控制烘干温度为60-70℃,烘干时间为6-8h。
本发明还提供了一种毛细管电色谱柱,包括石英毛细管柱以及位于石英毛细管柱内的固定相,固定相的制备原料包括上述氧化石墨烯-共价有机骨架材料复合材料。
具体的,毛细管电色谱柱的制备原料按质量份(质量份以mg为单位)或体积份(体积份以ml为单位)计包括:10-15质量份氧化石墨烯-共价有机骨架材料复合材料、5-10体积份反应试剂、50-80份质量份脱水剂、0.1-0.2质量份3-氨基丙基三乙氧基硅烷和10-15体积份分散溶剂。
在一种可能的设计中,脱水剂为二环己基碳二亚胺,分散溶剂为乙腈。
本发明还提供了一种氧化石墨烯-共价有机骨架材料毛细管电色谱柱的制备方法,包括以下步骤:
步骤a:将氧化石墨烯-共价有机骨架材料复合材料溶于反应试剂中,超声分散,得到第一混合液;
步骤b:在第一混合液中加入脱水剂和3-氨基丙基三乙氧基硅烷,超声充分溶解,静置后离心去除上层清液,得到黑色固体;
步骤c:黑色固体使用反应试剂清洗,烘干,得到3-氨基丙基三乙氧基硅烷修饰的氧化石墨烯-共价有机骨架材料复合材料;
步骤d:将石英毛细管柱活化,氮气吹干;
步骤e:将3-氨基丙基三乙氧基硅烷修饰氧化石墨烯-共价有机骨架材料复合材料分散在分散溶剂中,得到悬浊液,将悬浊液注入毛细管柱内静置,氮气吹干,静置,得到氧化石墨烯-共价有机骨架材料复合材料作为固定相的毛细管电色谱柱。
具体的,步骤a中,反应试剂为n,n-二甲基甲酰胺。
具体的,步骤a中,氧化石墨烯-共价有机骨架材料复合材料的质量与反应试剂的体积比过大会导致材料不能完全溶解;过小会浪费试剂。因此,控制氧化石墨烯-共价有机骨架材料复合材料的质量与反应试剂的体积比为10-15mg:5-10ml。
具体的,步骤a中,超声分散的温度过高会影响材料性质;温度过低会导致分散不完全。时间过长会导致温度升高;时间过短会导致分散不完全。因此,控制超声分散的温度为20-30℃;时间为20-40min。
具体的,步骤b中,为了保证复合材料和3-氨基丙基三乙氧基硅烷能够充分反应。因此,控制第一混合液、脱水剂和3-氨基丙基三乙氧基硅烷的质量体积比为:5-10ml(其中包含氧化石墨烯-共价有机骨架材料复合材料10-15mg,反应试剂5-10ml):50-80mg:0.1-0.2ml。
具体的,步骤b中,超声的温度过高会影响材料性质;温度过低会使材料之间反应不完全。时间过长会影响材料结构;时间过短会使反应不完全。因此,控制超声的温度为20-30℃;时间为10-30min;静置时间过长会破坏材料结构;时间过短会使反应不完全;因此,控制静置时间为10-12h。
具体的,步骤c中,烘干温度为60-70℃。
具体的,步骤d中的活化过程包括:先使用0.8-1.2mol/l氢氧化钠冲洗20-40min,再使用0.08-0.12mol/l盐酸冲洗5-15min,最后使用超纯水冲洗5-15min。
具体的,步骤e中,分散溶剂为乙腈,考虑到3-氨基丙基三乙氧基硅烷修饰氧化石墨烯-共价有机骨架材料复合材料在分散溶剂中的浓度过大会导致复合材料过多,使毛细管柱堵塞,过小会使进入毛细管柱的复合材料过少,影响分离效果。因此,控制3-氨基丙基三乙氧基硅烷修饰氧化石墨烯-共价有机骨架材料复合材料在分散溶剂中的浓度为1mg/ml。
具体的,步骤e中,静置的目的是保证复合材料充分键合在毛细管柱内壁。静置时间过长会导致浪费时间,时间过短可能会使材料在毛细管柱内壁不稳定,容易脱落。因此,控制静置时间为7-9h。
实施例1
本实施例提供了一种氧化石墨烯-共价有机骨架材料复合材料;氧化石墨烯-共价有机骨架材料复合材料的制备原料质量体积比为氧化石墨烯、反应溶剂和共价有机骨架材料单体为30mg:40ml:50mg。
上述氧化石墨烯-共价有机骨架材料复合材料的制备方法包括以下步骤:
步骤1:将30mg氧化石墨烯在20ml乙醇和四氢呋喃的混合液(乙醇和四氢呋喃的体积比为9:1)中超声30min使其分散均匀;
步骤2:加入10.5mg三醛基间苯三酚,超声15min充分溶解,得到第一混合悬浊液;
步骤3:将第一混合悬浊液离心5min,去除上清液,再加入15mg联苯二胺、0.1ml三氟乙酸和10ml反应溶剂进行超声反应10min得到第二混合悬浊液;
步骤4:将第二混合悬浊液离心5min,去除上清液,再加入10.5mg三醛基间苯三酚、15mg联苯二胺和0.1ml三氟乙酸,加入10ml反应溶剂进行超声反应60min,得到含有氧化石墨烯-共价有机骨架材料的第三混合悬浊液;
步骤5:将第三混合悬浊液冷却到室温,离心5min,去除上清液,清洗下层的氧化石墨烯-共价有机骨架材料,然后烘干6h,得到氧化石墨烯-共价有机骨架材料复合材料固体。
本实施例的技术方案工艺较为简单,制备总时间为8h10min,相比于现有处理技术,大大减少了制备时间、经济效益显著;本实施例的氧化石墨烯-共价有机骨架材料复合材料的扫描电镜图如图2所示。图3为tpbd型共价有机骨架材料的扫描电镜图,由图3可以看到tpbd型共价有机骨架呈海胆状,且团聚在一起。由图2可以看出,共价有机骨架材料均匀分布在了氧化石墨烯片上。合成的复合材料的比表面积为269m2/g。
实施例2
本实施例提供了一种毛细管电色谱柱;毛细管电色谱柱包括石英毛细管柱以及位于石英毛细管柱内的固定相,固定相的制备原料包括上述实施例1的氧化石墨烯-共价有机骨架材料复合材料。
图1所示为毛细管电色谱柱的制备方法示意图,毛细管电色谱柱的制备方法包括如下步骤:
步骤a:将10mg氧化石墨烯-共价有机骨架材料复合材料溶于5mln,n-二甲基甲酰胺中,超声20min使其分散,得到第一混合液;
步骤b:在5ml第一混合液中加入50mg脱水剂和0.1ml3-氨基丙基三乙氧基硅烷,超声10min充分溶解,静置10h后离心去除上层清液,得到黑色固体;
步骤c:黑色固体使用n,n-二甲基甲酰胺清洗,烘干(60℃),得到3-氨基丙基三乙氧基硅烷修饰的氧化石墨烯-共价有机骨架材料复合材料;
步骤d:将石英毛细管柱先使用0.8mol/l氢氧化钠冲洗30分钟,再使用0.12mol/l盐酸冲洗15min,最后使用超纯水冲洗15min,该过程是为了完成毛细管柱内壁的活化过程,使其表面带有硅氧基,之后氮气吹干;
步骤e:将3mg3-氨基丙基三乙氧基硅烷修饰氧化石墨烯-共价有机骨架材料复合材料分散在3ml乙腈中,得到悬浊液,将悬浊液注入毛细管柱内静置,氮气吹干,静置7h,得到氧化石墨烯-共价有机骨架材料复合材料作为固定相的毛细管电色谱柱。
本实施例制得的含有氧化石墨烯-共价有机骨架材料复合材料的毛细管电色谱柱的扫描电镜图如图5所示,本实施例的毛细管电色谱柱的内壁粗糙,能够很好的证明材料修饰到了毛细管柱内壁上。
对比例1
对比例1为普通毛细管柱。
对比例1的普通毛细管柱的扫描电镜图如图6所示,普通毛细管柱的内壁光滑,与修饰后的毛细管电色谱柱不同。通过对比图6和图5表明复合材料成功修饰入毛细管柱内壁上。
性能检测:
将上述实施例2制备的含有氧化石墨烯-共价有机骨架复合材料的毛细管电色谱柱在毛细管电色谱模式下用于磺胺类药物的分离,分离检测步骤操作如下:
(1)样品配制:预先配制3.0mg/ml的磺胺类药物标准溶液,分别取适量标准溶液混合稀释成浓度为30μg/ml磺胺类药物混合标准溶液,4℃冷藏备用;
(2)缓冲液的配制:配40mm磷酸盐储备液,使用磷酸调节ph至6.85-6.95,如果浓度和ph过高或过低,均会使分离度下降。使用水相过滤器过滤,4℃冷藏备用;
(3)分离检测:31.0cm的含有氧化石墨烯-共价有机骨架的毛细管电色谱柱,在10.0cm处烧检测窗口,装入卡盒中,使用毛细管电泳仪实现分离检测。进样量为0.5psi×5s,紫外检测波长为214nm。
本发明的实施例2的毛细管电色谱柱对十种磺胺类药物进行分析检测的色谱图如下图4所示;由图4可以看出十种磺胺类药物在4分钟内便可达到基线分离,且峰形良好,分离速度快,分离效率高。
表1为实施例2的毛细管电色谱柱与对比例1的普通毛细管柱对其中6种磺胺类药物分离性能参数对比。实施例2的毛细管电色谱柱的理论塔板数要明显高于普通毛细管柱,且分离度均能够达到1.5以上,实现基线分离。由此可见,本发明实施例的毛细管电色谱柱的性能优于普通毛细管柱。
表1实施例2、对比例1的分离性能结果
表2为实施例2的毛细管电色谱柱分离磺胺类药物的日内及日间精密度分析结果,由表2可以看出,十种磺胺类药物峰面积的日内相对标准偏差在0.33%-3.64%之间,日间精密度在1.65%-7.16%之间,表明实施例2的毛细管电色谱柱稳定性好。
表2实施例2的毛细管电色谱柱分离磺胺类药物的日内及日间精密度分析结果
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!