一种盐酸多奈哌齐口腔崩解片的质量控制方法与流程
本发明涉及一种药物制剂的质量控制方法,具体涉及一种盐酸多奈哌齐口腔崩解片的质量控制方法。
背景技术:
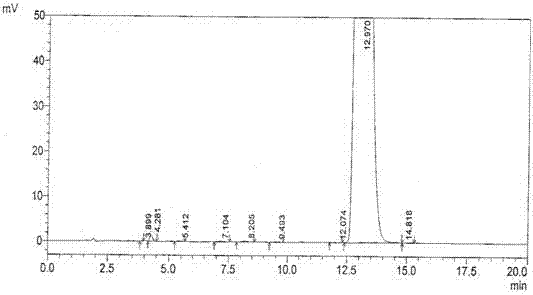
:口腔崩解片是近年来在分散片、咀嚼片的研究基础上发展的一种新剂型。随着世界范围内老龄化社会的来临,老年人在总人口中所占比例越来越多。随着年龄的增大,人体多种生理功能出现退化。老年人中多发的帕金森氏症、中风、老年性痴呆等疾病,都会导致手臂振颤或神经反射功能障碍等现象的发生,采用传统口服给药治疗可能会存在一定困难。口腔崩解片只需将片子放入口中,不需水或少量水,片刻后药物就随唾液到达吸收组织器官,为老年人安全服药提供了方便。盐酸多奈哌齐是一种对轻度或中度阿尔茨海默型痴呆症有较好治疗作用的药物,能较好的改善ad患者的认知功能障碍、精神行为异常和日常生活自理能力,减轻痴呆程度,且不良反应少、安全性高、耐受性好,现国内上市的仅有普通片和胶囊两种剂型,但盐酸多奈哌齐口腔崩解片在国外已经获得fda的批准上市。目前,国内外对于合成盐酸多奈哌齐的工艺较多,所用原料及合成路线也相差较大,得到的成品中大多含有较多的有机杂质成分。其中,本发明采用1-苄基-4-哌啶甲醛ⅰ和5,6-二甲氧基-1-茚酮ⅱ为原料,在nah催化作用下,经过aldol缩合反应制备盐酸多奈哌齐中间体ⅲ(盐酸多奈哌齐有关物质a),再将该盐酸多奈哌齐中间体ⅲ经过催化氢化得到多奈哌齐,最后经过盐酸成盐,得到盐酸多奈哌齐的合成路线中,2个起始原料中可能残留的为5,6-二甲氧基-1-茚酮ⅱ,而1-苄基-4-哌啶甲醛ⅰ为液体,可以完全参与合成反应,且易溶于乙酸乙酯、二氯甲烷等有机溶剂,在合成和精制过程中可以除尽,其余可能残留的副反应产物为氢化反应步骤产生的5,6-二甲氧基-2-(哌啶-4-甲基)-1-茚酮(脱苄基多奈哌齐),还可能残留的中间体为氢化不完全的盐酸多奈哌齐中间体(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮ⅲ,这是一种较重要的中间产物。美国药典(usp35)根据不同合成路线的盐酸多奈哌齐原料,使用了2种方法检查有机杂质,第一种方法检查了3个已知杂质——脱苄基多奈哌齐、羟基多奈哌齐、盐酸多奈哌齐中间体ⅲ(化合物ⅲ)、未知单杂和总杂质;第二种方法检查了5个已知杂质(脱苄基多奈哌齐、多奈哌齐吡啶类似物、苄基多奈哌齐溴代物、脱氢脱氧多奈哌齐、脱氧多奈哌齐)、未知单杂和总杂质。2种方法共检查7个已知杂质、未知单杂和总杂质,盐酸多奈哌齐口腔崩解片共检查3个已知杂质(脱苄基多奈哌齐、开环多奈哌齐、多奈哌齐-n-氧化物)、未知单杂和总杂质。其中盐酸多奈哌齐口腔崩解片检查的已知杂质中,1个杂质和原料一致(脱苄基多奈哌齐),另外2个已知杂质主要为氧化和过渡氧化的降解产物(开环多奈哌齐、多奈哌齐-n-氧化物),具体检测项目情况见表1所示:表1美国药典(usp35)盐酸多奈哌齐有关物质检查项目美国药典收载的本品降解产物主要为开环多奈哌齐、多奈哌齐-n-氧化物,其他副反应产物及中间杂质较多。目前国内关于盐酸多奈哌齐口腔崩解片的质量控制标准较少,其中对于中间杂质盐酸多奈哌齐有关物质a的质量检测及质量控制方法未制定相关标准。技术实现要素:为了解决现有技术的不足,本发明所要解决的技术问题是提供一种盐酸多奈哌齐口腔崩解片的质量控制方法。一种盐酸多奈哌齐口腔崩解片的质量控制方法,包括性状的观察、内容物的鉴别、含量均匀度的检查、溶出度的测定、崩解时限的测定、水分的测定、有关物质的测定和对含有的成分进行含量测定,其中所述对含有的成分进行含量测定包括盐酸多奈哌齐有关物质a的制备及其含量的测定,所述盐酸多奈哌齐有关物质a为(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,与美国药典收录的盐酸多奈哌齐口腔崩解片的质量标准相比较,本公司制剂增加了两项有关物质检测项(有关物质a和5,6-二甲基-1-茚酮),且部分有关物质检测控制限度高于美国药典标准,其中所述有关物质含量的测定包括以下步骤:a、取适量含盐酸多奈哌齐25mg的本品细粉,置50ml量瓶中,加5%甲醇溶液溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;量取1ml供试品溶液,置100ml量瓶中,加5%甲醇溶液稀释至刻度,摇匀,作为对照溶液;另取盐酸多奈哌齐、脱苄基多奈哌齐、盐酸多奈哌齐有关物质a、开环多奈哌齐、多奈哌齐-n-氧化物、5,6-二甲氧基-1-茚酮对照品各适量,置同一量瓶中,用甲醇溶解并稀释制成每1ml各含5μg的溶液,作为对照品溶液;以5%甲醇溶液作为空白溶剂对照溶液;b、照中国药典2015年版四部通则0512高效液相色谱法试验,取对照品溶液20μl,注入液相色谱仪,用十八烷基硅烷键合硅胶为填充剂,柱温35℃,流速1.4ml/min;按照以下体积比,以乙腈:3.85g/l癸烷磺酸钠溶液=35:65为流动相,用高氯酸调节癸烷磺酸钠溶液的ph为2.0,检测波长为271nm;理论板数按盐酸多奈哌齐计算不低于3000,盐酸多奈哌齐和相邻杂质峰的分离度应符合要求;c、取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%~20%;再精密量取空白溶剂对照溶液、供试品溶液、对照溶液、对照品溶液各20μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍;d、供试品溶液色谱图中如出现与对照品溶液色谱图中任一保留时间一致的色谱峰,按外标法以峰面积计算含量,每片含脱苄基多奈哌齐、开环多奈哌齐、多奈哌齐-n-氧化物分别不得大于盐酸多奈哌齐标示量的0.2%,盐酸多奈哌齐有关物质、5,6-二甲氧基-1-茚酮的含量分别不得大于盐酸多奈哌齐标示量的0.1%;其他任一单个未知杂质峰面积不得大于对照溶液主峰面积0.1%;各杂质峰面积的和不得大于对照溶液主峰面积1.0%。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述盐酸多奈哌齐有关物质a的合成路线为:其制备方法包括以下步骤:1)、称量:按照一定比例,取化合物ⅰ、化合物ⅱ和nah,待用;所述化合物ⅰ、化合物ⅱ与nah的摩尔比为1:1.02:1.2;2)、制备盐酸多奈哌齐中间体ⅲ的粗品:取无水四氢呋喃,通氮气,加nah,搅拌,加化合物ⅱ,加热条件下搅拌反应;再滴加化合物ⅰ,继续搅拌反应,反应完后缓慢滴加一定量的水,将得到的反应液干燥,加碳酸氢钠水溶液,搅拌,过滤,取滤饼,水洗滤饼,干燥,得化合物ⅲ粗品;所述无水四氢呋喃是由四氢呋喃与无水硫酸钠除水处理而得;按照1mol化合物ⅱ:4000ml碳酸氢钠水溶液的量加入碳酸氢钠水溶液,所述碳酸氢钠水溶液的浓度为11%;3)、第一次精制:取b步骤得到的化合物ⅲ粗品,加乙酸乙酯,加热溶解,加活性炭,加热回流搅拌,过滤,降温搅拌析晶,过滤,取滤饼,用乙酸乙酯洗涤,干燥,得化合物ⅲ精制品;4)、第二次精制:向c步骤得到的化合物ⅲ精制品中加乙酸乙酯,加热溶解,过滤,降温搅拌析晶,过滤,取滤饼,用乙酸乙酯洗涤,干燥,得化合物ⅲ。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述盐酸多奈哌齐有关物质a的有关物质的测定包括以下步骤:s1:取本品,精密称定,加流动相溶解并定量稀释制成每1ml中含100μg的溶液,作为供试品溶液;精密量取适量,用流动相定量稀释制成每1ml中含1.0μg的溶液,作为对照溶液;另取5,6-二甲氧基-1-茚酮适量,加流动相溶解并定量稀释制成每1ml中含1.0μg的溶液,作为对照品溶液;s2:照中国药典2015年版四部通则0512高效液相色谱法试验,用十八烷基硅烷键合硅胶为填充剂,以0.2mol/l乙酸钠:9%乙酸:乙腈按照体积比25:30:45混合液为流动相,所述流动相用三乙胺调节ph值为7.0;检测波长271nm;理论板数按盐酸多奈哌齐有关物质计算不低于3000,盐酸多奈哌齐有关物质和相邻杂质峰的分离度应符合要求;s3:取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%~20%;再精密量取供试品溶液、对照溶液、对照品溶液各20μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的的2倍;s4:供试品溶液色谱图中如出现与对照品溶液色谱图中5,6-二甲氧基-1-茚酮峰保留时间一致的色谱峰,按外标法以峰面积计算,其含量不得大于0.1%;其他任一单个未知杂质峰面积不得大于对照溶液主峰面积1/10;各杂质峰面积的和不得大于对照溶液主峰面积1.0%。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述盐酸多奈哌齐有关物质a的含量测定包括以下步骤:取0.2g盐酸多奈哌齐有关物质a,精密称定,加冰醋酸30ml溶解后,加醋酐20ml,滴加结晶紫指示剂1滴,照电位滴定法(中国药典2015年版四部通则0701),用高氯酸滴定液(0.1mol/l)滴定,并将滴定的结果用空白试验校正。每1ml高氯酸滴定液(0.1mol/l)相当于37.75mg的c24h27no3。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述性状的观察为本品为白色或类白色片。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述内容物的鉴别为包括以下步骤:(1)取含量测定项下的供试品溶液,加水制成每1ml含20μg的溶液,按照中国药典2015年版四部通则0401紫外-可见分光光度法测定,在271nm与315nm的波长处有最大吸收;(2)取本品细粉适量,加水使盐酸多奈哌齐溶解,过滤,滤液显中国药典2015年版四部通则0301收录的氯化物的鉴别反应;(3)在含量测定项下记录的色谱图中,供试品的保留时间应与对照品主峰的保留时间一致。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述含量均匀度的检查为:取本品1片,置250ml量瓶中,加水稀释至刻度,摇匀,滤过,取续滤液,按照中国药典2015年版四部通则0401紫外-可见分光光度法,在315nm的波长处测定吸收度;另取在105℃下干燥至恒重盐酸多奈哌齐对照品适量,精密称定,加水制成每1ml含20μg的溶液,同法测定,并计算每片的含量,应符合中国药典2015年版四部通则0941的规定。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述溶出度的测定为取本品,照中国药典2015年版四部通则0931第二法记录的溶出度测定方法,以0.1mol/l的盐酸溶液900ml为溶出介质,转速每分钟50转,依法操作,经15分钟时,取溶液适量,滤过,取续滤液作为供试品溶液。另精密称取盐酸多奈哌齐对照品适量,用0.1mol/l的盐酸溶液溶解并稀释制成每1ml约含5.5μg的溶液,作为对照品溶液;精密量取对照品溶液和供试品溶液各20μl,照含量测定项下的色谱条件测定,计算每片的溶出量,限度为标示量的90%,应符合规定。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述崩解时限的测定为取本品6片,照中国药典2015年版四部通则0921崩解时限检查法测定,各片均应在1分钟内全部崩解。上述一种盐酸多奈哌齐口腔崩解片的质量控制方法,其中所述水分的测定为取本品,照中国药典2015年版四部通则0832第一法水分测定法测定,含水分不得超过5.0%。本发明具有如下有益效果:本发明合成盐酸多奈哌齐路线中,不会产生美国药典(usp35)收载的盐酸多奈哌齐的其他副反应产物或中间体,且本发明制定了盐酸多奈哌齐口腔崩解片的质量控制方法,以及研究了杂质盐酸多奈哌齐有关物质a的制备方法和质量控制方法,进一步提高了盐酸多奈哌齐口腔崩解片的质量,完善了现有行业标准,具有简单易行,精确度和准确性高等优点,对提高产品质量具有重要意义。附图说明图1盐酸多奈哌齐混合对照品溶液高效液相色谱图;图2盐酸多奈哌齐口腔崩解片供试品溶液高效液相色谱图;图3盐酸多奈哌齐有关物质a红外吸收光谱图;图4盐酸多奈哌齐有关物质ax射线粉末衍射谱;图5盐酸多奈哌齐有关物质a差示扫描量热分析(dsc);图6盐酸多奈哌齐有关物质a热重(tga)谱;图7盐酸多奈哌齐有关物质a高分辨质谱(hrms)谱;图8盐酸多奈哌齐有关物质a紫外吸收光谱图。具体实施方式下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实施例范围之中。实施例1盐酸多奈哌齐口腔崩解片的质量控制一、本发明一种盐酸多奈哌齐口腔崩解片的质量控制方法1、有关物质含量的测定包括以下步骤:a、取适量含盐酸多奈哌齐25mg的本品细粉,置50ml量瓶中,加5%甲醇溶液溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;量取1ml供试品溶液,置100ml量瓶中,加5%甲醇溶液稀释至刻度,摇匀,作为对照溶液;另取盐酸多奈哌齐、脱苄基多奈哌齐、盐酸多奈哌齐有关物质a、开环多奈哌齐、多奈哌齐-n-氧化物、5,6-二甲氧基-1-茚酮对照品各适量,置同一量瓶中,用甲醇溶解并稀释制成每1ml各含5μg的溶液,作为对照品溶液;以5%甲醇溶液作为空白溶剂对照溶液;b、照中国药典2015年版四部通则0512高效液相色谱法试验,取对照品溶液20μl,注入液相色谱仪,用十八烷基硅烷键合硅胶为填充剂,柱温35℃,流速1.4ml/min;按照以下体积比,以乙腈:3.85g/l癸烷磺酸钠溶液(用高氯酸调节ph为2.0)=35:65为流动相,检测波长为271nm;理论板数按盐酸多奈哌齐计算不低于3000,盐酸多奈哌齐和相邻杂质峰的分离度应符合要求;c、取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%~20%;再精密量取空白溶剂对照溶液、供试品溶液、对照溶液、对照品溶液各20μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍;d、供试品溶液色谱图中如出现与对照品溶液色谱图中任一保留时间一致的色谱峰,按外标法以峰面积计算含量,每片含脱苄基多奈哌齐、开环多奈哌齐、多奈哌齐-n-氧化物分别不得大于盐酸多奈哌齐标示量的0.2%,盐酸多奈哌齐有关物质、5,6-二甲氧基-1-茚酮的含量分别不得大于盐酸多奈哌齐标示量的0.1%;其他任一单个未知杂质峰面积不得大于对照溶液主峰面积1/10;各杂质峰面积的和不得大于对照溶液主峰面积1.0%。表2盐酸多奈哌齐口腔崩解片有关物质与美国药典检验标准对比2、性状的观察本品外观为白色或类白色片。3、内容物的鉴别包括以下步骤:(1)取含量测定项下的供试品溶液,加水制成每1ml含20μg的溶液,按照中国药典2015年版四部通则0401紫外-可见分光光度法测定,在271nm与315nm的波长处有最大吸收;(2)取本品细粉适量,加水使盐酸多奈哌齐溶解,过滤,滤液显中国药典2015年版四部通则0301收录的氯化物的鉴别反应;(3)在含量测定项下记录的色谱图中,供试品的保留时间应与对照品主峰的保留时间一致。4、含量均匀度的检查取本品1片,置250ml量瓶中,加水稀释至刻度,摇匀,滤过,取滤液,按照中国药典2015年版四部通则0401紫外-可见分光光度法,在315nm的波长处测定吸收度;另取在105℃下干燥至恒重盐酸多奈哌齐对照品适量,精密称定,加水制成每1ml含20μg的溶液,同法测定,并计算每片的含量,应符合中国药典2015年版四部通则0941的规定。5、溶出度的测定方法:取本品,照中国药典2015年版四部通则0931第二法记录的溶出度测定方法,以0.1mol/l的盐酸溶液900ml为溶出介质,转速每分钟50转,依法操作,经15分钟时,取溶液适量,滤过,取续滤液作为供试品溶液。另精密称取盐酸多奈哌齐对照品适量,用0.1mol/l的盐酸溶液溶解并稀释制成每1ml约含5.5μg的溶液,作为对照品溶液;精密量取对照品溶液和供试品溶液各20μl,照含量测定项下的色谱条件测定(盐酸多奈哌齐混合对照品溶液高效液相色谱图见图1所示;盐酸多奈哌齐口腔崩解片供试品溶液高效液相色谱图见图2所示),计算每片的溶出量,限度为标示量的90%,应符合规定。表3盐酸多奈哌齐口腔崩解片溶出度测定含量结果批号15分钟时溶出度(%)30分钟时溶出度(%)100201101.198.2100202100.7100.3100203105.4105.7市售品121205a102.299.76、崩解时限的测定取本品6片,照中国药典2015年版四部通则0921崩解时限检查法测定,各片均应在1分钟内全部崩解。中试三批样品及市售产品(安理申)1批对比检查,中试三批样品均符合规定(见表4)。表4盐酸多奈哌齐口腔崩解片崩解时限检查结果7、水分的测定本品为快速崩解片,片剂的硬度相对普通片小一些,为防止吸湿裂片,需对水分严格控制,在本品质量标准中增加水分检查,即取本品,照水分测定法(中国药典2015年版四部通则0832第一法)测定,根据三批中试样品及市售产品测定结果,控制限度为不得过5.0%,中间体控制水分在4.0%以内(本发明产品试验结果见下表5)。表5盐酸多奈哌齐口腔崩解片水分测定结果实施例2盐酸多奈哌齐有关物质a的制备按照以下步骤,共制备得到三批次(即20140201、20140202和20140203)盐酸多奈哌齐有关物质a——(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮ⅲ,制备工艺如下,制备结果见表6所示:(1)盐酸多奈哌齐有关物质a粗品制备在2000ml棕色反应瓶中加入无水四氢呋喃800ml(按起始原料1mol:4000ml计算,100ml四氢呋喃需使用2g无水硫酸钠除水处理,过滤),加入nah5.76g(0.24mol),搅拌使其悬浮,并在溶液中通入氮气。加入5,6-二甲氧基-1-茚酮39.2g(0.204mol),持续氮气保护,并加热搅拌反应。使用恒压漏斗滴加1-苄基-4-哌啶甲醛40.6g(0.2mol),滴加完毕后,持续氮气保护,在加热条件下继续搅拌反应。缓慢滴加约10ml水终止反应。反应液减压蒸干,加400ml1%碳酸氢钠水溶液(按起始原料1mol:4000ml计算)搅拌洗涤,过滤,滤饼水洗涤至洗脱液几乎无色。滤饼减压干燥4h,得(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮的粗品,黄色固体65.8g。粗品收率为87.3%。(2)盐酸多奈哌齐有关物质a第一次精制取(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮的粗品10g,加乙酸乙酯200ml,加热溶解,加入活性炭,加热回流搅拌吸附,趁热过滤。滤液降温搅拌析晶2h,过滤,滤饼用乙酸乙酯共洗涤3次。滤饼减压干燥4h,得(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮精制品ⅰ,65.8g粗品共获得类白色固体53.6g。第一次精制收率为81.5%。(3)盐酸多奈哌齐有关物质a第二次精制取(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮的粗品10g加乙酸乙酯200ml,加热溶解完全,趁热过滤。滤液降温搅拌析晶2h,过滤,滤饼用乙酸乙酯洗涤3次。滤饼减压干燥4h,得(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮的精制品ⅱ,53.6g精制品ⅰ共获得白色固体48.5g,第二次精制收率为90.5%。按理论投料量0.2mol计算总收率为64.3%。表6盐酸多奈哌齐有关物质a三批样品制备结果实施例3盐酸多奈哌齐有关物质a的质量控制一、盐酸多奈哌齐有关物质a的有关物质的测定包括以下步骤:s1:取本品,精密称定,加流动相溶解并定量稀释制成每1ml中含100μg的溶液,作为供试品溶液;精密量取适量,用流动相定量稀释制成每1ml中含1.0μg的溶液,作为对照溶液;另取5,6-二甲氧基-1-茚酮适量,加流动相溶解并定量稀释制成每1ml中含1.0μg的溶液,作为对照品溶液;s2:照中国药典2015年版四部通则0512高效液相色谱法试验,用十八烷基硅烷键合硅胶为填充剂,按照体积比,以0.2mol/l乙酸钠:9%乙酸:乙腈按照体积比25:30:45混合后的混合液为流动相,所述流动相用三乙胺调节ph值为7.0;检测波长271nm;理论板数按盐酸多奈哌齐有关物质计算不低于3000,盐酸多奈哌齐有关物质和相邻杂质峰的分离度应符合要求;s3:取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%~20%;再精密量取供试品溶液、对照溶液、对照品溶液各20μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的的2倍;s4:供试品溶液色谱图中如出现与对照品溶液色谱图中5,6-二甲氧基-1-茚酮峰保留时间一致的色谱峰,按外标法以峰面积计算,其含量不得大于0.1%;其他任一单个未知杂质峰面积不得大于对照溶液主峰面积1/10;各杂质峰面积的和不得大于对照溶液主峰面积1.0%。二、盐酸多奈哌齐有关物质a含量检测1、杂质分析及含量检测方法盐酸多奈哌齐有关物质aⅲ合成路线仅一步反应,主要有关物质为起始原料5,6-二甲氧基-1-茚酮,1-苄基-4-哌啶甲醛(液体)完全参与合成反应,且易溶于乙酸乙酯、二氯甲烷等有机溶剂,在合成和精制过程中可以除尽。取本品约0.2g,精密称定,加冰醋酸30ml溶解后,加醋酐20ml,滴加结晶紫指示剂1滴,照电位滴定法(中国药典2015年版四部通则0701),用高氯酸滴定液(0.1mol/l)滴定,并将滴定的结果用空白试验校正。每1ml高氯酸滴定液(0.1mol/l)相当于37.75mg的c24h27no3;同时按照上述方法配置空白溶液(除了不加本发明盐酸多奈哌齐有关物质a外,其余均一样),检测空白的干扰情况。结果显示,空白溶液对方法无干扰。2、三批样品的含量测定分别精密称取生产的三批盐酸多奈哌齐有关物质a(ⅲ)样品,按拟定的盐酸多奈哌齐有关物质a(ⅲ)含量测定方法检测。结果盐酸多奈哌齐有关物质a(ⅲ)的含量均符合规定,盐酸多奈哌齐有关物质a(ⅲ)的含量限度拟定为95.0%~105.0%(见表7)。表7三批盐酸多奈哌齐有关物质a样品的含量测定三、盐酸多奈哌齐有关物质a的结构确证1、红外吸收光谱测试单位:四川大学华西药学院测试仪器:bruker公司vector22型红外吸收光谱仪测试方法:kbr压片测试结果:(批号:20140201)见表8及附图3所示:表8盐酸多奈哌齐有关物质a红外吸收光谱解析:(1)3449cm-1:-oh伸缩振动;1077cm-1:=c-o伸缩振动;1689cm-1:c=o伸缩振动,所以样品中含有羰基、烯醇结构。(2)3075cm-1为=c-h的伸缩振动峰,同时在1646cm-1处存在c=c的伸缩振动峰,所以样品中含有双键结构。(3)3030cm-1为苯环的=c-h伸缩振动峰,855cm-1为苯环四取代的=c-h弯曲振动峰,735cm-1以及697cm-1处吸收,为苯环单取代的=c-h弯曲振动峰,所以样品中存在单取代苯环和四取代苯环。1603cm-1和1499cm-1为苯环骨架的c=c伸缩振动峰。(4)1396cm-1以及1368cm-1处的吸收峰为叔丁基的c-h弯曲振动峰,所以样品中存在叔丁基结构。(5)1215cm-1为c-n伸缩振动峰,所以样品中有c-n的存在。(6)2929cm-1、2798cm-1为c-h伸缩振动峰,所以样品中存在-ch3和-ch2的结构。由红外图谱可知,样品(批号20140201)的官能团与多奈哌齐中间体(ⅲ)的官能团基本符合,所以样品(批号20140201)即(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮。由红外图谱可知,该样品中同时存在少量烯醇式的异构体。由样品(批号:20140201)的红外光谱特征吸收峰可知,样品的结构信息与多奈哌齐有关物质a结构中主要官能团符合。2、粉末衍射(xrpd)测试单位:四川大学分析测试中心测试仪器:x’pertprompdx射线衍射仪测试数据:(批号20140201)(见附图4)结论:由测试结果可知,本品为结晶性粉末。3、差示扫描量热分析(dsc)测试单位:中科院成都分院分析测试中心仪器型号:dscq200v24.2build107测试条件:吹扫气:高纯n2;吹扫速度100ml/min;升温速度:10℃/min;温度范围:25~190℃测试结果:(批号:20140201)(见表9及附图5)表9盐酸多奈哌齐有关物质a热分析dsc数据样品编号吸热起始温度(℃)吸热峰温(℃)20140201177.83178.82dsc结论:本品dsc谱显示一个较尖锐的吸热峰,熔点约178.82℃。4、热重(tga)测试单位:中科院成都分院分析测试中心仪器型号:tgaq500v20.10build36测试条件:吹扫气:高纯n2,吹扫速度100ml/min;升温速度:10℃/min;温度范围室温(10℃)—400℃测试结果:(批号:20140201)(见表10及附图6)表10盐酸多奈哌齐有关物质a热分析tga数据样品编号初始分解温度范围(℃)失重(%)20140201281.5391.48tga结论:由tga数据可知,本品在温度低于200℃时,无重量损失,因此本品不含结晶水,温度在220℃以上是重量开始损失,281.53℃为本品初始分解温度,重量损失91.48%。5、高分辨质谱(hrms)测试单位:中国科分院成都生物所离子化方式:esi测试仪器:microtof-qii10203测试结果:(批号:20140201)(见表11及附图7)表11盐酸多奈哌齐有关物质a高分辨质谱数据及结构式结论:本品高分辨质谱显示,m+1峰分子量为378.2058,同理论m+1峰对应的分子式为c24h28no3,计算分子量378.2064相差1.6ppm。高分辨率质谱报告指出:盐酸多奈哌齐有关物质a的分子式为:c24h27no3;结构式为:综上所述,样品(批号20140201)即为多奈哌齐有关物质a(e)-2-[(1-苄基哌啶-4-基)亚甲基]-5,6-二甲氧基-1-茚酮(化合物ⅲ),c24h27no3,相对分子质量377.48,含c24h27no3不得少于99.0%。四、盐酸多奈哌齐有关物质a质量标准的研究①外观本品三批试制产品外观均为白色结晶性粉末。因此质量标准拟定为“本品为白色结晶性粉末”。本品在水中不溶,在甲醇极微溶解,在乙酸乙酯中微溶,在二氯甲烷中溶解。熔点本品的熔点(中国药典2015年版四部通则0612)为176℃~178℃。②溶解性取本品适量,精密称定,分别采用水、乙酸乙酯、甲醇、二氯甲烷作为溶剂,考察其溶解性,结果质量标准拟定为“本品在水中不溶,在甲醇、乙酸乙酯中微溶,在二氯甲烷中溶解。”③熔点仪器型号:yrt-3熔点测定仪生产厂家:天大天发科技有限公司取本品,依法检查熔点(中国药典2015年版四部通则0612),结果表明本品熔点为176℃~178℃,与文献报道一致。因此本品质量标准拟定为“熔点(中国药典2015年版四部通则0612)为176℃~178℃”。紫外光谱鉴别:取盐酸多奈哌齐有关物质a,加甲醇溶解并稀释制成1ml中含10μg的溶液,照紫外-可见分光光度法(中国药典2015年版四部通则0401)测定,在200-400nm波长范围内扫描。结果本品最大吸收波长为339.60nm、286.80nm、225.20nmnm,最小吸收波长为308.50nm、241.00nm、220.00nm(见附图8)。因此质量标准拟定为“取本品适量,加甲醇溶解并稀释制成每1ml中约含10μg的溶液,照中国药典2015年版四部通则0401紫外-可见分光光度法测定,在338nm、287nm、225nm的波长处有最大吸收。干燥失重:取本品,在105℃干燥至恒重,减失重量不得超过1.0%(中国药典2015年版四部通则0831)。具体为取本品,在105℃干燥至恒重,减失重量不得超过0.5%(中国药典2015年版四部通则0831)。三批盐酸多奈哌齐有关物质a测定结果均符合规定(见表12)。表12盐酸多奈哌齐有关物质a紫外吸收试验结果批号干燥失重%201402010.080201402020.087201402030.087含量测定:取本品约0.2g,精密称定,加冰醋酸30ml溶解后,加醋酐20ml,滴加结晶紫指示剂1滴,照电位滴定法(中国药典2015年版四部通则0701),用高氯酸滴定液(0.1mol/l)滴定,并将滴定的结果用空白试验校正。每1ml高氯酸滴定液(0.1mol/l)相当于37.75mg的c24h27no3。贮藏:遮光,密封,室温干燥处保存。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1