贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法及检测方法与流程
本发明涉及医药领域,特别涉及贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法及检测方法。
背景技术:
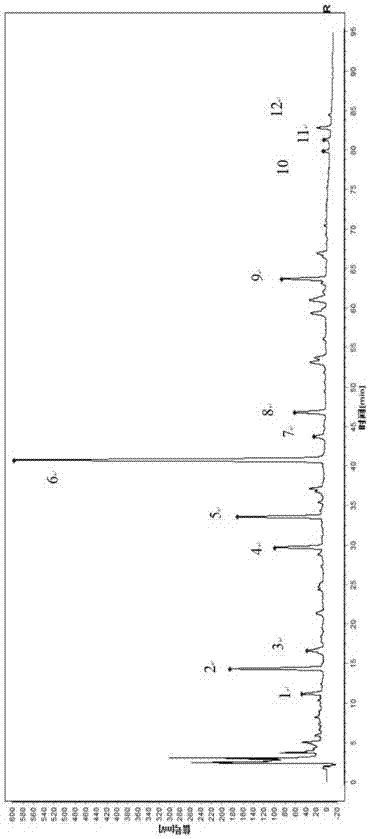
贞芪扶正颗粒由女贞子及黄芪两味药材组成,收载于《中药成方制剂》(第20册),具有提高人体免疫力,保护骨髓及肾上腺皮质的功能。大量的药学研究和临床应用已经证明其可作为癌症辅助放化疗的“明星”药物。目前,主要是采用处方中药味进行鉴别或者增加对某种单一成分进行含量测定的方法来提高贞芪扶正颗粒的质量标准。用一个或者两个成分来说明贞芪扶正颗粒的内在质量,具有一定的片面性。要控制贞芪扶正颗粒的质量,只针对其中一两个化学成分进行表征和控制是不够的,必须对它的物质群整体予以控制。现行质量标准仅对黄芪药材中黄芪甲苷进行定性及定量分析,未对女贞子中成分进行限定,中药复方成分的复杂性为其质量控制增加了难度。所以,除了微观分析外,还应该用某种宏观分析的方法,从整体上有效的表征中药质量。指纹图谱作为中药及其制剂的质量控制方法,目前已经成为业内人士的共识。但是以蒸发光检测器条件下建立的指纹图谱受检测器本身的影响因素较大,建立的指纹图谱相似度较差;有些技术虽建立紫外检测器条件下的指纹图谱,但是图谱分离基线较差。因此有必要建立特征图谱或指纹图谱对该制剂质量的一致性及稳定性进行评价。技术实现要素:有鉴于此,本发明提供一种贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法及检测方法。该方法采用高效液相色谱法建立贞芪扶正颗粒的中间体特征图谱及成品指纹图谱,用于贞芪扶正颗粒的质量控制。为了实现上述发明目的,本发明提供以下技术方案:本发明提供了一种贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法,包括如下步骤:步骤1:取贞芪扶正颗粒的中间体或贞芪扶正颗粒制备供试品溶液;步骤2:获得对照品溶液;步骤3:分别取所述供试品溶液和所述对照品溶液经高效液相色谱法测定,获得贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱;所述中间体的制备方法为:取黄芪、女贞子加水煎煮,过滤,收集滤液浓缩至70~80℃时密度为1.10~1.30的浸膏。所述中间体或成品中的活性成分为毛蕊异黄酮葡萄糖苷、芒柄花素、红景天苷和/或特女贞苷;贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱分别包括12个特征峰,其中,2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号为芒柄花素。步骤1与步骤2的顺序不分先后。在本发明的一些具体实施方案中,贞芪扶正颗粒的中间体特征图谱的构建方法中,供试品溶液的制备方法为:取所述浸膏与水混合,用水饱和正丁醇振摇提取,合并正丁醇液,蒸干,残渣加甲醇溶解,定容,过滤,收集滤液。在本发明的一些具体实施方案中,贞芪扶正颗粒的中间体特征图谱的构建方法中,供试品溶液的制备方法为:取浸膏约0.5-5.0g,精密称定,加水5-100ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取2-5次,每次10-80ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至1-100ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。在本发明的一些具体实施方案中,贞芪扶正颗粒指纹图谱的构建方法中,供试品溶液的制备方法为:取贞芪扶正颗粒与水混合,用水饱和正丁醇振摇提取,合并正丁醇液,蒸干,残渣加甲醇溶解,定容,过滤,收集滤液。在本发明的一些具体实施方案中,贞芪扶正颗粒指纹图谱的构建方法中,供试品溶液的制备方法为:取贞芪扶正颗粒约1.0-10.0g,研细,精密称定,加水10-100ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取2-5次,每次10-80ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至1-100ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。在本发明的一些具体实施方案中,贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法中,所述对照品溶液的制备方法为:取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品,与甲醇混合制得分别含芒柄花素、红景天苷、毛蕊异黄酮葡萄糖苷、特女贞苷的溶液。在本发明的一些具体实施方案中,所述对照品溶液的制备方法为:取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品适量,精密称定,加甲醇制成每1ml分别含芒柄花素50μg、红景天苷45μg、毛蕊异黄酮葡萄糖苷45μg、特女贞苷50μg的溶液,即得。在本发明的一些具体实施方案中,所述高效液相色谱法测定的色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b;梯度洗脱过程如下:在本发明的一些具体实施方案中,所述高效液相色谱法测定的检测波长为200~280nm,柱温为20~40℃,流速为0.5~1.5ml/min;理论板数按特女贞苷计算应不低于2000。在本发明的一些具体实施方案中,所述贞芪扶正颗粒的中间体特征图谱包括12个特征峰,其中,2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号为芒柄花素。在本发明的一些具体实施方案中,所述贞芪扶正颗粒的中间体特征图谱与对照品峰对应的峰为s峰,获得各特征峰与s峰的相对保留时间,所述相对保留时间应在规定值的±5%之内。作为优选,供试品特征图谱中应呈现12个特征峰,其中4个峰应分别与相应的对照品峰保留时间相同;与特女贞苷对照品峰相应的峰为s峰,计算其余特征峰的相对保留时间,其相对保留时间应在规定值的±5%之内。所述规定值为:1号峰为0.28、2号峰为0.35、3号峰为0.41、4号峰为0.73、5号峰为0.82、6号峰为1.00、7号峰为1.07、8号峰为1.15、9号峰为1.56、10号峰为1.97、11号峰为2.00、12号峰为2.04。在本发明的一些具体实施方案中,所述贞芪扶正颗粒指纹图谱包括12个特征峰,其中,2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号为芒柄花素。在上述技术方案的基础上,本发明还提供了贞芪扶正颗粒的检测方法,按照所述的构建方法获得贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱,对待测样品的中间体特征图谱与所述贞芪扶正颗粒的中间体特征图谱进行比较,相对保留时间应在规定值的±5%之内为合格产品;对待测样品的指纹图谱与所述贞芪扶正颗粒指纹图谱进行相似性评价,相似度大于0.90为合格产品。本发明以贞芪扶正颗粒中间体(黄芪、女贞子加水煎煮,滤过,滤液浓缩后的浸膏)及成品为研究对象,利用指纹图谱技术开展质量标准提升研究。其中,浓缩中间体是产品生产过程的关键质控部位,生产工艺中要求浓缩至密度1.10-1.30,生产按此执行,批间中间体为浸膏,存在密度差异,使单位质量浸膏中成分因密度异同存在含量差异,因此不适合以指纹图谱进行监测,适合建立特征图谱。本研究选择毛蕊异黄酮葡萄糖苷、芒柄花素、红景天苷和特女贞苷为特征图谱或指纹图谱参照物色谱峰,并建立特征图谱或指纹图谱,纳入企业内控质量标准,提高企业对贞芪扶正颗粒产品质量的监测水平,以期为完善该制剂的质量控制方法提供参考依据。本发明提供了一种贞芪扶正颗粒中间体特征图谱及成品指纹图谱检测方法及其色谱图谱,通过此方法可以较为全面地控制贞芪扶正颗粒的质量,从而更好地保证贞芪扶正颗粒的质量稳定性、一致性和可控性,从而确保贞芪扶正颗粒的安全性和有效性。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。图1示对照特征图谱;其中,12个特征峰中峰2:红景天苷;峰5:毛蕊异黄酮葡萄糖苷;峰6(s):特女贞苷;峰10:芒柄花素;图2示混合对照品色谱图;其中,2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号为芒柄花素;图3示10批次中间体共有模式叠加色谱图;图4示对照指纹图谱;其中,12个共有峰中,峰2:红景天苷;峰5:毛蕊异黄酮葡萄糖苷;峰6(s):特女贞苷;峰10:芒柄花素;图5示混合对照品的hplc色谱图;其中,2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号峰为芒柄花素;图6示10批次贞芪扶正颗粒共有模式叠加色谱图。具体实施方式本发明公开了一种贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法及检测方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本
发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本发明提供了一种贞芪扶正颗粒中间体特征图谱构建方法,包括以下步骤:(1)供试品溶液的制备取浸膏约0.5-5.0g,精密称定,加水5-100ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取2-5次,每次10-80ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至1-100ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。(2)参照物溶液的制备取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品适量,精密称定,加甲醇制成每1ml分别含芒柄花素50μg、红景天苷45μg、毛蕊异黄酮葡萄糖苷45μg、特女贞苷50μg的溶液,即得。(3)测定:精密吸取参照物溶液及供试品溶液各5μl,注入液相色谱仪,测定,记录95分钟的色谱图,即得。作为优选,所述高效液相色谱测定的色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按下表中的规定进行梯度洗脱;检测波长为200-280nm,柱温为20-40℃,流速为0.5-1.5ml/min。理论板数按特女贞苷计算应不低于2000。作为优选,梯度洗脱过程如下本发明还提供了一种贞芪扶正颗粒成品指纹图谱构建方法,包括以下步骤:(1)供试品溶液的制备取本品约1.0-10.0g,研细,精密称定,加水10-100ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取2-5次,每次10-80ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至1-100ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。(2)参照物溶液的制备取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品适量,精密称定,加甲醇制成每1ml分别含芒柄花素50μg、红景天苷45μg、毛蕊异黄酮葡萄糖苷45μg、特女贞苷50μg的溶液,即得。(3)测定:精密吸取参照物溶液及供试品溶液各5μl,注入液相色谱仪,测定,记录95分钟的色谱图,即得。作为优选,所述高效液相色谱测定的色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按下表中的规定进行梯度洗脱;检测波长为200-280nm,柱温为20-40℃,流速为0.5-1.5ml/min。理论板数按特女贞苷计算应不低于2000。作为优选,梯度洗脱过程如下本发明提供了一种贞芪扶正颗粒中间体特征图谱及成品指纹图谱检测方法及其色谱图谱,通过此方法可以较为全面地控制芪扶正颗粒的质量,从而更好地保证芪扶正颗粒的质量稳定性、一致性和可控性,从而确保芪扶正颗粒的安全性和有效性。本发明提供的贞芪扶正颗粒的中间体特征图谱或贞芪扶正颗粒指纹图谱的构建方法及检测方法中所用原料及试剂均可由市场购得。下面结合实施例,进一步阐述本发明:实施例1中间体特征图谱照高效液相色谱法(《中国药典》2015版四部通则0512)测定。色谱条件与系统适应性实验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按下表中的规定进行梯度洗脱;检测波长为220nm,柱温为30℃,流速为1.0ml/min。理论板数按特女贞苷计算应不低于2000。表1时间(分钟)流动相a(%)流动相b(%)0~85→895→928~158→1192→8915~3011→1889→8230~4518→2182→7945~5221→2379→7752~6523→2877→7265~7528→3772→6375~8537→5063→5085~955050对照品溶液的制备取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品适量,精密称定,加甲醇制成每1ml分别含芒柄花素50μg、红景天苷45μg、毛蕊异黄酮葡萄糖苷45μg、特女贞苷50μg的溶液,即得。供试品溶液的制备取浸膏约1.0g,精密称定,加水10ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取3次,每次20ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。测定法精密吸取对照品溶液及供试品溶液各5μl,注入液相色谱仪,测定,记录95分钟的色谱图(如图1所示),即得。供试品特征图谱中应呈现12个特征峰,其中4个峰应分别与相应的对照品峰保留时间相同;与特女贞苷对照品峰相应的峰为s峰,计算其余特征峰的相对保留时间,其相对保留时间应在规定值的±5%之内。规定值为:0.28(峰1)、0.35(峰2)、0.41(峰3)、0.73(峰4)、0.82(峰5)、1.00(峰s)、1.07(峰7)、1.15(峰8)、1.56(峰9)、1.97(峰10)、2.00(峰11)、2.04(峰12)。实施例2中间体特征图谱的建立特征图谱共有模式的建立及部分特征峰的指认按照供试品溶液的制备方法,制备10批次中间体的供试品溶液,按照上述色谱条件进样5μl,测定图谱。将10批次样品的图谱导入国家药典委员会的“中药色谱指纹图谱相似度评价系统软件(2012版)”,生成特征图谱共有模式,标定12个特征峰,通过与对照品保留时间对比分析,确定2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号为芒柄花素。10批次中间体特征图谱相对保留时间比较采用平均矢量法对10批次中间体谱图建立共有模式,并计算相对保留时间,得到各图谱与对照图谱的相对保留时间分析表,分析结果见表2,因此,确定特征峰的相对保留时间规定值为0.28(峰1)、0.35(峰2)、0.41(峰3)、0.73(峰4)、0.82(峰5)、1.00(峰s)、1.07(峰7)、1.15(峰8)、1.56(峰9)、1.97(峰10)、2.00(峰11)、2.04(峰12)。表210批中间体特征图谱相对保留时间分析表10批次中间体特征图谱相似度比较采用平均矢量法对10批次贞芪扶正颗粒中间体特征图谱建立共有模式,将每张特征图谱与对照特征图谱进行比较,并计算各个样本与基准样本的夹角余弦和相关系数,得到各图谱与对照特征图谱的相似度,分析结果见表3。10批次贞芪扶正颗粒中间体中各成分特征图谱的相似度良好,均在0.90以上,符合要求。表310批中间体特征图谱相似度分析表实施例3成品指纹图谱照高效液相色谱法(《中国药典》2015版四部通则0512)测定。色谱条件与系统适用性实验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按下表中的规定进行梯度洗脱;检测波长为220nm,柱温为30℃,流速为1.0ml/min。理论板数按特女贞苷计算应不低于2000。表4时间(分钟)流动相a(%)流动相b(%)0~85→895→928~158→1192→8915~3011→1889→8230~4518→2182→7945~5221→2379→7752~6523→2877→7265~7528→3772→6375~8537→5063→5085~955050对照品溶液的制备取芒柄花素对照品、红景天苷对照品、毛蕊异黄酮葡萄糖苷对照品和特女贞苷对照品适量,精密称定,加甲醇制成每1ml分别含芒柄花素50μg、红景天苷45μg、毛蕊异黄酮葡萄糖苷45μg、特女贞苷50μg的溶液,即得。供试品溶液的制备取本品约15.0g,研细,精密称定,加水40ml使溶解,置分液漏斗中,用水饱和正丁醇振摇提取3次,每次20ml,合并正丁醇液,蒸干,残渣加甲醇溶解并转移至5ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。测定法精密吸取对照品溶液及供试品溶液各5μl,注入液相色谱仪,测定,记录95分钟的色谱图(如图5所示),即得。按中药色谱指纹图谱相似度评价系统,供试品指纹图谱与对照指纹图谱经相似度计算,相似度不得低于0.90。实施例4贞芪扶正颗粒指纹图谱的建立贞芪扶正颗粒指纹图谱共有模式的建立及部分共有峰的指认按照供试品溶液的制备方法,制备10批次贞芪扶正颗粒的供试品溶液,按照上述色谱条件进样5μl,测定指纹图谱。将10批次样品的图谱导入国家药典委员会的“中药色谱指纹图谱相似度评价系统软件(2012版)”,生成指纹图谱共有模式,共有峰12个。通过与对照品保留时间对比分析,确定2号峰为红景天苷,5号峰为毛蕊异黄酮葡萄糖苷,6号峰为特女贞苷,10号峰为芒柄花素。表510批次贞芪扶正颗粒共有峰相对保留时间表610批次贞芪扶正颗粒共有峰相对峰面积10批次贞芪扶正颗粒成品指纹图谱相似度比较采用平均矢量法对10批次贞芪扶正颗粒成品指纹图建立共有模式,将每张指纹图谱与对照指纹图谱进行比较,并计算各个样本与基准样本的夹角余弦和相关系数,得到各图谱与对照指纹图谱的相似度,分析结果见表7。10批次贞芪扶正颗粒中各成分指纹图谱的相似度良好,均在0.90以上,符合要求。表710批次贞芪扶正颗粒指纹图谱相似度以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1