人类短片段串联重复序列高通量测序信息的处理方法与流程
本发明涉及生物检测领域,特别涉及一种人类短片段串联重复序列高通量测序信息的处理方法。
背景技术:
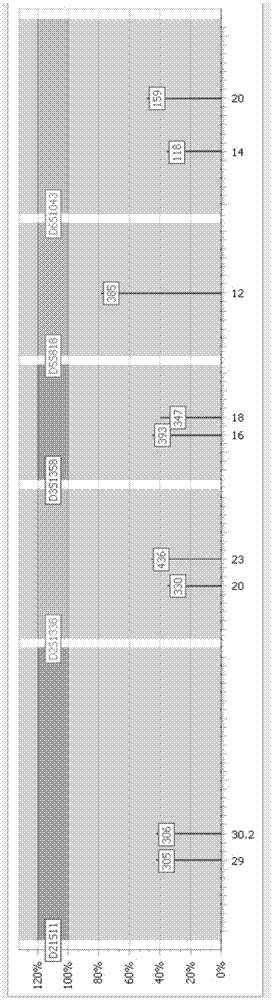
:短片段串联重复序列(shorttandemrepeat,简称str)是广泛存在于人类染色体dna中的一类多态性遗传标记系统,因其存在范围广(平均16kb中即有一个str基因座),核心序列小(2-7bp)且扩增产物长度均小于500bp,等位基因位点的数字即代表序列重复的次数。str基因座的等位基因片断长度集中,故可对多个str基因座进行复合扩增。复合扩增多个str基因座,累计鉴别能力可以接近或达到dna指纹水平,是现代法医学使用范围最广的dna指纹标记。自上世纪90年代以来,对str通用的检测方法是以多重pcr检测约20个基因座的基因型,在检测中使用以荧光标记的引物并设计好扩增子的长度,使所产生的不同长短的具有荧光标记的针对每个基因座的扩增子在毛细管电泳中分离,并与标准物进行比对,从而实现对每个基因座中的等位基因进行分型。但是,这种方法也存在着由于技术上的限制而带来的缺陷,主要有:(1)由于荧光标记物的相互干扰和毛细管长度及成像技术等方面的限制,被分析基因座的数目已难以进一步大幅提升;(2)由于分析的对象是各个片段的长度大小,无法进一步检测到组成片段的核酸一级结构的微小差异,因此限制了检测的分辨度;(3)出峰宽度受电泳条件影响,导致碱基个数相差1-2bp时难易分辨。而高通量测序法的出现则能够弥补以上缺陷,其具有以下特点:(1)检测位点数几乎不受平台限制;(2)核心重复数一致的情况下,测定出的序列微变异可以进一步区分不同个体,提高检测的分辨度;(3)序列信息直接反映核心重复数,更加准确。此外,各测序公司已经开展应用高通量测序法平台测定人类str基因座的研究工作,包括罗氏的gsflx、illumina的gaiix和lifetechnology的pgm平台。商业化的str高通量测序法检测试剂盒已开始逐渐涌现,其中不乏国产试剂盒。然而,发明人发现,通过高通量测序法得到的高通量测序信息,其信息量非常大,基于此,有必要对人类短片段串联重复序列高通量测序信息进行简单快速地处理,以得到常规的str分型结果,同时,将其中不同人个体的str序列的微变异以直观的方式显现出来。技术实现要素:本发明实施例所要解决的技术问题在于,提供了一种人类短片段串联重复序列高通量测序信息的处理方法。具体技术方案如下:人类短片段串联重复序列高通量测序信息的处理方法,所述处理方法包括:步骤a、获取单张芯片的str高通量测序信息作为原始序列,并根据预设测序长度,过滤所述原始序列,保留具有所述预设测序长度的序列,形成第一待处理序列;步骤b、根据样本标签信息,将所述第一待处理序列分类至不同的样本文件夹中,然后根据str目的片段特异引物信息,分别将每个所述样本文件夹中的第一待处理序列再分类至不同的str基因座文件夹中,以在每个所述str基因座文件夹中形成第二待处理序列;步骤c、建立针对不同str基因座的阶梯参比序列,作为比对基础数据库,将每个所述str基因座文件夹中的所述第二待处理序列分别与所述比对基础数据库中相对应str基因座的序列进行比对,在每个所述str基因座文件夹中保留序列相似度大于等于90%的序列,形成第三待处理序列;步骤d、将样本测序条目数的第一阈值设定为1000,将基因座测序条目数的第二阈值设定为50,将基因座内分型测序条目数的第三阈值设定为5,将基因座内分型测序条目数/基因座测序条目数的第四阈值设定为40%,对每个所述str基因座文件夹中的所述第三待处理序列进行筛选,在每个所述str基因座文件夹中获取同时大于等于第一阈值、第二阈值、第三阈值和第四阈值的序列,得到str分型结果。进一步地,所述处理方法还包括:建立不同的亚型阶梯参比序列,所述不同的亚型阶梯参比序列具有相同str核心区重复数,同时具有不同的str序列结构;根据所述亚型阶梯参比序列的命名规律,将每个所述str基因座文件夹中获取的str分型结果进一步分型至不同的亚型,形成分型图形树,所述分型图形树包括不同的基因座、每个所述基因座的分型及分型数量、每个所述基因座的分型的亚型及亚型数量;将所述分型图形树转化成excel表格,通过合并所述excel表格,以获取每个所述基因座的分型及分型数量,通过展开所述excel表格,以获取每个所述基因座的分型、每个所述基因座的分型的亚型及亚型数量。具体地,所述分型数量包括:每种分型的个数以及所述每种分型在所述基因座中所占的比例,以每种分型在所述基因座中所占比例作为图形化展示纵坐标,平衡由于pcr扩增效率不同导致的基因座之间的分型数量差异;所述亚型数量包括:每种亚型的个数以及所述每种亚型在所述基因座中所占的比例,以每种亚型在所述基因座中所占比例作为图形化展示纵坐标,平衡由于pcr扩增效率不同导致的基因座之间的亚型数量差异。具体地,所述比对基础数据库通过获取已公开的序列信息以及通过实测大量样本的序列信息而建立。作为优选,所述比对基础数据库中的序列信息为可调整的。具体地,作为优选,所述步骤a还包括:在对所述原始序列进行过滤时,获取所述原始序列的长度分布图。具体地,作为优选,所述步骤b还包括:在将所述第一待处理序列分类至不同的样本文件夹中时,获取所述第一待处理序列的标签类别分布图。具体地,作为优选,所述步骤b还包括:在将每个所述样本文件夹中的第一待处理序列再分类至不同的str基因座文件夹中时,获取所述第一待处理序列的基因座类别分布图。具体地,所述str分型结果包括:不同的样本标签类别、每种样本标签类别中所包含的基因座类别、每种基因座类别中所包含的等位基因信息。作为优选,所述步骤a至所述步骤d支持一键式操作;可选地,所述处理方法还包括多序列比对,即将所述第三待处理序列分成多个子序列,将每个所述子序列与所述比对基础数据库中相对应str基因座的序列进行比对,获取每个所述子序列的序列突变信息,并以图形化展示突变信息。本发明实施例提供的技术方案带来的有益效果是:本发明实施例提供的人类短片段串联重复序列高通量测序信息的处理方法,通过对芯片的原始str高通量测序信息依次进行长度筛选、样本分类、基因座分类、序列比对、比对结果筛选,可将原始的人类短片段串联重复序列高通量测序信息快速有效地转换成str分型结果,实现高通量测序结果和现有技术常用的str报告格式之间的无缝对接。可见,本发明实施例提供的处理方法可快速处理高信息量的高通量测序信息,有效提高了str的检测速率及检测精度,对于生物检测具有积极的意义。附图说明为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1-1是本发明实施例提供的示例性的原始序列的长度分布示意图;图1-2是本发明又一实施例提供的示例性的原始序列中全部子序列的长度分布示意图;图2是本发明又一实施例提供的示例性的第一待处理序列的标签类别分布示意图;图3是本发明又一实施例提供的示例性的第一待处理序列的基因座类别分布示意图;图4-1是本发明又一实施例提供的,对d21s11、d2s1338、d3s1358、d5s818、d6s1043中各自所含的第三待处理序列进行筛选之前,其各自的序列示意图;图4-2是本发明又一实施例提供的,对d21s11、d2s1338、d3s1358、d5s818、d6s1043中各自所含的第三待处理序列进行筛选之后,其各自的序列示意图;图5是本发明又一实施例提供的分型图形树的示意图;图6是本发明又一实施例提供的在进行序列突变图形展示过程中所形成的图形化界面示意图。具体实施方式除非另有定义,本发明实施例所用的所有技术术语均具有与本领域技术人员通常理解的相同的含义。为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明实施方式作进一步地详细描述。本发明实施例提供了一种人类短片段串联重复序列高通量测序信息的处理方法,该处理方法包括以下步骤:步骤101、获取单张芯片的str高通量测序信息作为原始序列,并根据预设测序长度,过滤原始序列,保留具有预设测序长度的序列,形成第一待处理序列。可以理解的是,步骤101中所述的预设测序长度可以根据实际测序需求来确定,其可以为测序目的片段的读长下限(如60bp等),如此可提高str分型结果的针对性。此外,本发明实施例提供的处理方法可在计算机终端设备上基于与其对应的软件来实现,在进行步骤101之前,可以对每一次的处理过程进行新建项目,例如包括项目命名以及保存地址确认等。进一步地,为了更直观地观测原始序列的长度分布,步骤101还包括:在对原始序列进行过滤时,获取原始序列的长度分布图。举例来说,该原始序列的长度分布图可以包括:表示具有预设测序长度的序列(即长度合格序列)和小于预设测序长度的序列(即长度不合格序列)所占比例的第一分布图,其如图1-1所示。该原始序列的长度分布图还可以包括:原始序列中全部子序列的长度分布,其如附图1-2所示,其中附图1-2中,横坐标表示序列长度,单位为bp,纵坐标表示子序列的个数。在步骤101中对原始序列进行完长度筛选以获取第一待处理序列之后,本发明实施例将进行步骤102以对第一待处理序列进行分类处理。具体地,步骤102如下所示:根据样本标签信息,将第一待处理序列分类至不同的样本文件夹中,然后根据str目的片段特异引物信息,分别将每个样本文件夹中的第一待处理序列再分类至不同的str基因座文件夹中,以在每个str基因座文件夹中形成第二待处理序列。其中,每个样本标签对应有不同的str核心序列,其优选使用阿拉伯数字进行顺序编号。举例来说,样本标签的信息可以如表1所示:表1样本标签编号str核心序列code001ctaaggtagccode002aagaggaatccode003cagatggaaccode004ttggtgattccode005ttccgagaatcode006tgagtggatccode007tcacgaatacode008tacgtggtaccode009tctatcggatcode010ttggagtctc进一步地,在将第一待处理序列分类至不同的样本文件夹中时,获取第一待处理序列的标签类别分布图,其示例如图2所示,可见,每个样本文件夹的命名通过样本标签信息及其相对应的序列信息来确定。通过提供标签类别分布图,可以更加直观地浏览第一待处理序列中所含的str核心序列。在将第一待处理序列分类成不同的样本文件夹之后,本发明实施例还根据str目的片段特异引物信息,将每个样本文件夹中的第一待处理序列再分类至不同的str基因座文件夹中,以在每个str基因座文件夹中形成第二待处理序列。其中,上述的str目的片段特异引物信息根据测序目的片段的序列和pcr引物设计原则所确定,其所代表的意义是一小段寡聚的dna,一般有两个(一对),分为上游引物和下游引物,分别指导dna两条链的聚合。它们主要作用有两个,一个是和模板特异性结合来指导taq聚合酶合成所要的片段。一个是提供一个3’端的-oh末端,只有拥有一个-oh末端,dna聚合酶才能合成dna。进一步地,步骤102还包括:在将每个样本文件夹中的第一待处理序列再分类至不同的str基因座文件夹中时,获取第一待处理序列的基因座类别分布图,其示例如图3所示,可见,每个str基因座文件夹的命名通过基因座信息以及其所占第一待处理序列的比例所确定。步骤103、建立针对不同str基因座的阶梯参比序列,作为比对基础数据库,将每个str基因座文件夹中的第二待处理序列分别与比对基础数据库中相对应str基因座的序列进行比对,在每个str基因座文件夹中保留序列相似度大于等于90%的序列,形成第三待处理序列。具体地,上述比对基础数据库通过获取已公开的序列信息以及通过实测大量样本的序列信息而建立。可见,通过对先有技术公开的信息进行检索来获取已公开的序列信息,同时通过对大量样本(例如为3000-5000或者更多个样本)进行实测以获取它们的序列信息并将此作为比对基础,可提高后续str分型结果的精确度。上述比对基础数据库并不是一成不变的,即比对基础数据库中的序列信息为可调整的,当有新的序列信息被发现时,或者其内已含有的序列信息不适用时,可以对比对基础数据库进行实时更新,并以更新后的序列信息作为比对基础,如此不仅保证该比对基础数据库内的序列信息更加全面,且保证了分型结果的精确度。此外,为了提高str分型结果的准确度,优选在每个str基因座文件夹中保留序列相似度大于等于95%,例如96%、97%、98%的序列,以形成第三待处理序列。可以理解的是,当在计算机中基于特定的软件平台来实施本发明实施例提供的处理方法时,可以将该比对基础数据库预置在软件中。在上述比对基础数据库中可以分成多个数据区,以针对每种不同的基因座形成一个完整的比对数据库。以str基因座d21s11举例来说,针对该基因座的阶梯参比序列如表2所示:表2分型核心区参比序列28a[tcta]4[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1028b[tcta]5[tctg]6[tcta]3tatcta]3tca[tcta]2tccata[tcta]928#1[tcta]6[tctg]5[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]928#2[tcta]5[tctg]5[tcta]3ta[tcta]2tca[tcta]2tccata[tcta]1128#3[tcta]6[tctg]5[tcta]3ta[tcta]3tca[tcta]tccata[tcta]1028.2a[tcta]4[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]9ta[tcta]28.2b[tcta]5[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]8ta[tcta]29a[tcta]4[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1129b[tcta]6[tctg]5[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1029#1[tcta]7[tctg]5[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]929#2[tcta]5[tctg]5[tcta]3ta[tcta]2tca[tcta]2tccata[tcta]1229.2#1[tcta]5[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]9ta[tcta]30#1[tcta]7[tctg]5[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1030#2[tcta]6[tctg]4[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1230a[tcta]6[tctg]5[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1130b[tcta]5[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1130c[tcta]4[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]1230d[tcta]6[tctg]6[tcta]3ta[tcta]3tca[tcta]2tccata[tcta]10在表2中,以诸如a、b、c、d等字母作为后缀的参比序列是经过对已公开的序列信息进行检索而得到;以#作为后缀的参比序列是通过样本实测而得到的。步骤104、将样本测序条目数的第一阈值设定为1000,将基因座测序条目数的第二阈值设定为50,将基因座内分型测序条目数的第三阈值设定为5,将基因座内分型测序条目数/基因座测序条目数的第四阈值设定为40%,对每个str基因座文件夹中的第三待处理序列进行筛选,在每个str基因座文件夹中获取同时大于等于第一阈值、第二阈值、第三阈值和第四阈值的序列,得到str分型结果。具体地,在步骤104中,样本测序条目数指的是具有相同的特定样本标签信息的第三待处理序列,即位于同一样本文件夹目录下的所有第三待处理序列;基因座测序条目数指的是同一样本文件夹下,某str基因座文件夹中的所有第三待处理序列。基因座内分型测序条目数指的是同一样本文件夹下,某str基因座文件夹内与该str基因座某参阶梯参比序列相似度大于等于90%的第三待处理序列。举例来说,可以按如下所示的方法进行上述各步骤:1)获取单张芯片的str高通量测序信息作为原始序列(3,014,265条);2)根据预设测序长度,过滤所述原始序列,保留具有所述预设测序长度(例如≧60bp)的序列,形成第一待处理序列(例如为2,842,460条);3)根据样本标签信息和str目的片段特异引物信息,逐级将第一待处理序列分类至84个样本文件夹目录下的16个不同str基因座文件夹中,形成第二待处理序列(条目总数仍为2,842,460条,分至1344个文件夹);4)将每个str基因座文件夹中的第二待处理序列(平均每个文件夹约含2115个测序条目)分别与比对基础数据库中相对应str基因座的阶梯参比序列进行比对,在每个所述str基因座文件夹中保留序列相似度大于等于90%的序列,形成第三待处理序列(条目总数降为2,201,691条,平均每个str基因座文件夹含1638个测序条目);根据如下参数和阈值(示例见表3)进行筛选并获得分型结果。表3参数意义软件内参数显示名称阈值样本测序条目数barcodereads总数≧1000基因座测序条目数基因座reads总数≧50基因座内分型测序条目数基因分型reads数≧5分型测序条目数/基因座测序条目数%基因分型%≧40%进一步如图4-1及图4-2所示例的,当对str基因座文件夹,例如d21s11、d2s1338、d3s1358、d5s818、d6s1043来说,当对它们其中各自所含的第三待处理序列进行筛选之前,其序列示意图如图4-1所示,而对它们其中各自所含的第三待处理序列进行筛选之后,其序列示意图如图4-2所示。可见,本领域技术人员可能够直观且快速地从图4-2中观察得到所需的str分型结果。相应地,当对d21s11、d2s1338、d3s1358、d5s818、d6s1043中的第三待处理序列进行筛选之后,所得到的str分型结果可如表4所示。表4样本标签基因座等位基因1等位基因2001d21s112930.2001d2s13382023001d3s13581618001d5s8181212001d6s10431420如表4可知,本发明实施例所得到的str分型结果可以包括,但不限于:不同的样本标签类别、每种样本标签类别中所包含的基因座类别、每种基因座类别中所包含的等位基因信息。进一步地,本发明实施例提供的处理方法还包括对步骤104所得str分型再分型至各亚型的以下步骤:步骤105、建立不同的亚型阶梯参比序列,不同的亚型阶梯参比序列具有相同str核心区重复数,同时具有不同的str序列结构。其中,上述亚型阶梯参比序列也是基于步骤103所建立的比对基础数据库来获取的,不同的亚型阶梯参比序列指的是亚型不同,但是处在同一阶梯,它们具有相同str核心区重复数,同时具有不同的str序列结构。可以理解的是,每个特定位点的str均由2部分构成:中间的核心区和外围的侧翼区。举例来说,以基因座d21s11举例来说,其所对应的亚型阶梯参比序列如表5所示;表5步骤106、根据亚型阶梯参比序列的命名规律,将每个str基因座文件夹中获取的str分型结果进一步分型至不同的亚型,形成分型图形树。分型图形树包括不同的基因座、每个基因座的分型及分型数量、每个基因座的分型的亚型及亚型数量。通过步骤106生成的分型图形树的示例可参见图5,可见,分型图形树可直观地展示每个str分型的亚型分型结果。可以理解的是,上述的命名规律指的是以数字+后缀(字母或#数字)来命名亚型阶梯参比序列,其中数字表示str核心区重复数,即str分型。后缀表示重复数相同情况下的不同亚型,已公开报道的采用字母做后缀,未报道的采用#数字做后缀;上述的“不同的基因座”可以理解为不同基因座的类别。步骤107、将分型图形树转化成excel表格,通过合并excel表格,以获取每个基因座的分型及分型数量,通过展开excel表格,以获取每个基因座的分型、每个基因座的分型的亚型及亚型数量。其中,上述的分型数量包括:每种分型的个数以及每种分型在基因座中所占的比例;亚型数量包括:每种亚型的个数以及每种亚型在基因座中所占的比例。同样地,以基因座d21s11举例来说,通过合并excel表格,针对该d21s11基因座的分型及分型数量如表6所示:表6进一步地,在表6的基础上,继续以基因座d21s11举例来说,通过表6,可获取针对该d21s11基因座的分型的亚型及亚型数量,如表7所示:表7本发明实施例可通过合并与展开的放置,对表6和表7所示的内容之间进行自由切换,以实现分型结果和亚型分型结果之间自由切换,举例来说,该切换示例可如表8所示:表8比对统计表格形式分型结果亚型分型结果合并29,30.2无展开29,30.229b,30.2a由上述可知,本发明实施例提供的方法通过设置具有相同str核心区重复数,但str序列不同的亚型阶梯参比序列,并根据其命名规律生成分型树形,实现str分型结果及其亚型分型结果之间的自由切换,如此即可使最后的结果报告的详细程度与高通量测序的优势相一致,且这种形式也便于未来不同个体间的str结果比对。进一步地,本发明实施例提供的处理方法还包括鉴定序列突变的步骤106:进行多序列比对,该多序列比对的过程如下:将步骤103中经过序列比对获取的第三待处理序列分成多个子序列,将每个子序列与比对基础数据库中相对应str基因座的序列进行比对,获取每个子序列的序列突变信息。进一步地,本发明实施例还可选地包括对序列突变进行图形化展示,以获取str分型结果中的序列微变异。本发明实施例可通过多序列比对和图形化界面相结合,直观展示str扩增产物区间内序列变异。首先确定多序列比对范围:多序列比对在和相同“阶梯参比序列”比中的测序条目中进行。例如编号为barcode92的样本测序结果中,共计347条和d13s317的12型参比序列比中,319条和d13s317的8型参比序列比中,多序列比对将分别在这两组测序结果和相应的参比序列范围内进行。再次,在进行完上述步骤之后,再进行序列突变图形展示,其图形化界面示例如图6所示。可见,通过观察图6,可容易地识别出其中的变异序列,从而直观地展示str扩增产物区间内序列变异。在对分型数量或亚型数量进行图形化展示的过程中,以每种分型或亚型在所述基因座中所占比例作为做纵坐标,以平衡由于pcr扩增效率不同导致的基因座之间的分型数量差异。可见,本发明实施例通过划定若干小范围的多序列比对,同时结合图形化界面,可快速直观地展示str范围内的序列变异,如此可有效提高个体认定、亲缘关系判断等法庭科学工作的效率和准确性。以上所述仅为本发明的较佳实施例,并不用以限制本发明的保护范围,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种构建Hi‑C高通量测序文库的方法与制造工艺
- 桑葚病原菌高通量鉴定及种属分类方法及其应用与制造工艺
- 基于高通量测序技术检测乳腺癌/卵巢癌易感基因的多重PCR特异性引物、试剂盒及方法与制造工艺
- 基于高通量测序检测人EGFR基因变异的质控方法及试剂盒与制造工艺
- 基于高通量测序技术的小麦病原微生物检测方法及其应用与制造工艺
- 一种基于二代高通量测序技术的地中海贫血症的基因检测试剂盒的制造方法与工艺
- 一种基于集群的高通量数据分析方法与制造工艺
- 高通量双折射干涉成像光谱仪装置及其成像方法与制造工艺
- 高通量测序检测人循环肿瘤DNA EGFR基因的捕获探针及试剂盒的制造方法与工艺
- 一种高通量测序检测无创亲子鉴定的方法与制造工艺