一种超支化聚合物及其制备方法和应用与流程
本发明涉及药剂制备技术领域,具体涉及一种磁共振成像造影用的造影剂及制备方法与应用。
背景技术:
随着科技的发展,磁共振成像已成为临床检测疾病的一种有效手段。相较于其它临床成像技术,磁共振成像在揭示解剖学结构,尤其是在探测炎症组织以及固体肿瘤方面具有诸多优势。此外磁共振成像还具有无电离辐射损伤、多参数成像为医生提供大量的诊断信息以及三维定位能力。但为了进一步提高成像的灵敏度和准确率,通常需要造影剂来增强成像对比度。
目前,临床用的造影剂一般为钆的小分子螯合物。这类小分子造影剂存在一些缺点,如血液循环时间短、弛豫率低、无靶向性,具有一定的毒性,临床上对于肾功能不全患者存在引起肾源性系统性纤维化的风险。因此制备高弛豫率、生物相容性好以及具有组织或肿瘤靶向性的mri造影剂成为发展的焦点。
近年来,多种不同的载体材料包括高分子、脂质体、胶束、无机或杂化纳米粒子被用于构建mri造影剂。其中将钆基小分子造影剂共价或者非共价的与高分子结合可以有效的提高对钆的负载率。并且将钆基小分子造影剂修饰到大分子骨架上,可以增强其体内稳定性、降低分子的旋转速率、提高弛豫效率、延长血液循环时间和在组织中的驻留时间,另外高分子具有较多的活性官能团,可对其进行靶向化学修饰,增强对组织或器官的靶向性;将高分子修饰为电中性,可以使渗透压与血浆相近,从而降低毒副作用。但多数合成高分子在体内排泄较慢或不彻底,限制了其在临床中的应用。因此,寻找生物相容性好以及可降解的大分子作为小分子造影剂的载体尤为重要。
技术实现要素:
本发明的主要目的在于提供一种具有生物相容性好、易生物降解和高弛豫率等特点的超支化聚赖氨酸为载体的mri造影剂及其制备方法,以克服现有技术中的不足,实现肿瘤的早期筛查。
这种超支化聚合物的制备方法,包括如下步骤:
超支化聚赖氨酸骨架的制备:将赖氨酸盐酸盐与苛性碱按照摩尔比为100:80~90的比例,在保护气氛下、140~160℃反应40~50h获得呈树枝状交联的超支化聚赖氨酸骨架;
第一超支化聚赖氨酸分子的制备:将所述超支化聚赖氨酸骨架与二乙烯三胺五乙酸按照质量比至少为1:5的比例在水中混合,在室温下反应6~8h获得偶联有二乙烯三胺五乙酸基的所述第一超支化聚赖氨酸分子;
第二超支化聚赖氨酸分子的制备:将所述第一超支化聚赖氨酸分子与钆化物在水中混合,在37~42℃反应6~8h后获得络合有钆离子的所述第二超支化聚赖氨酸分子;其中所述第一超支化聚赖氨酸分子与所述钆化物的质量比为1:1~1:2;
将所述第二超支化聚赖氨酸分子与叶酸活性酯在ph值为8~10的碳酸盐缓冲液中混合反应5~12h获得所述超支化聚合物;其中,所述第二超支化聚赖氨酸分子与所述叶酸的质量比10:1~10:5。
其中,所述第一超支化聚赖氨酸分子的制备步骤还包括加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐用于活化所述二乙烯三胺五乙酸中的羧基,所述1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐与所述二乙烯三胺五乙酸的摩尔比为5~6:1。
其中,所述第一超支化聚赖氨酸分子的制备步骤和所述第二超支化聚赖氨酸分子的制备步骤中,加入苛性碱或生物碱之一调节ph值为5.5~6.5。
其中,所述苛性碱为naoh或koh任一种;所述生物碱为四甲基乙二胺。
其中,所述超支化聚赖氨酸骨架的制备步骤、所述第一超支化聚赖氨酸分子的制备步骤、所述所第二超支化聚赖氨酸分子的制备步骤中均通过透析和冻干后分离的方法分别使所述超支化聚赖氨酸骨架、所述第一超支化聚赖氨酸分子、所述第二超支化聚赖氨酸分子与其他杂质分离。所述超支化聚合物的制备步骤中通过超滤后冻干分离的方法使所述超支化聚合物与杂质分离。
其中,所述钆化物为六水合氯化钆。
本发明提供的超支化聚合物,具有超支化聚赖氨酸骨架结构,所述超支化聚赖氨酸骨架结构式如式1所示:
其中,任一或多个第一末端-nh2上的一个h原子被r基团取代,所述r基团为络合gd3+的二乙烯三胺五乙酸基;
其中,任一或多个未连接有所述r基团的第二末端-nh2上的一个h原子被叶酸基取代。
本发明还提供这种超支化聚合物作为造影剂的应用。
其中,所述造影剂中含有如权利要求7所述超支化聚合物,其中gd3+浓度为80~100毫摩尔每升。
有益效果:
本发明获得超支化聚赖氨酸为载体的mri造影剂采用聚赖氨酸作为造影剂主体,赖氨酸是人体中本身就存在的氨基酸之一,具有生物相容性好,生物毒性低和生物可降解的优点。将钆螯合物连接在超支化聚赖氨酸末端氨基上,能够增加造影剂分子的尺寸,增加旋转相关时间,从而提高弛豫效率,并延长其血液循环时间,并且超支化聚赖氨酸末端密集的氨基实现了对钆的富集,经实验发现,该造影剂弛豫率(13.44mm-1·s-1)明显高于小分子造影剂的弛豫率(4.3mm-1·s-1)。此外,在大分子造影剂上修饰靶向基团可以提高造影剂的肿瘤靶向性。
与现有技术相比,本发明的有益效果包括:该超支化聚赖氨酸为载体的mri造影剂的生物相容性好,毒性低,弛豫率高,可从体内代谢,具有肿瘤靶向性,且其制备方法简单,可大量制备。
附图说明
图1是本发明造影剂与gd-dtpa弛豫率对比图;
图2是本发明造影剂与gd-dtpa的t1加权成像对比图;
图3是本发明造影剂与gd-dtpa在kb细胞中的细胞毒性测试图;
图4是本发明造影剂在293t细胞中的细胞毒性测试图;
图5是本发明造影剂在不同钆离子浓度下的无胸腺裸鼠上的组织毒理测试图;
图6是本发明造影剂在kb细胞中的细胞摄取图;
图7是本发明造影剂在移植kb细胞肿瘤的无胸腺裸鼠上的体内成像图。
具体实施方式
下面,将结合附图对本发明实施例做详细介绍。
本发明提供一种超支化聚赖氨酸为载体的mri造影剂的制备方法,包括如下步骤:
步骤一:将赖氨酸盐酸盐与氢氧化钾在研钵中充分研磨成浆状后,以液体石蜡油为导热媒介、在氮气保护气氛下、在150℃下反应48h,获得主要产物超支化聚赖氨酸骨架,以及副产物和其他杂质。
其中,所述氢氧化钾的投料量为赖氨酸盐酸盐摩尔数的90%。
步骤一的反应结束后缓冷到室温。倒掉液体石蜡,用石油醚反复洗,然后减压蒸除石油醚,得到棕色固体。所得棕色固体溶解在甲醇中,副产物氯化钾形成白色沉淀可通过过滤移除,获得的滤液备存。所述滤液转移至四氢呋喃中得到棕黄色沉淀物。过滤分离得到所述棕黄色沉淀物溶于蒸馏水中进行透析(截留分子量为3500),经过透析后获得透析液,冻干透析液,分离得到所述超支化聚赖氨酸骨架(hbpll),其化学结构式如式1所示:
这种超支化聚赖氨酸骨架是一种由多官能团的赖氨酸单体高度支化制备的聚合物。超支化聚合物中主要是支化部分,支化点较多,其具有一定的相对分子质量分布,与树枝形聚合物相似。这类聚合物分子具有类似球形的紧凑结构,流体力学回转半径小,分子链缠结少,所以相对分子质量的增加对粘度影响较小,而且分子中带有许多官能性端基,对其进行修饰可以改善其在各类溶剂中的溶解性,或得到功能材料,与线性同系物相比都具有较高的溶解性和较低的粘度。
步骤二:将所述超支化聚赖氨酸骨架溶解在水中,控制超支化聚赖氨酸骨架浓度不低于50mg/ml;然后加入投料量为超支化聚赖氨酸骨架质量的5倍的二乙烯三胺五乙酸(dtpa),形成第一反应液。通过生物碱四甲基乙二胺调节第一反应液ph值到6左右,然后加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc)至上述第一反应液中活化二乙烯三胺五乙酸的羧基,在室温下反应6h,使得超支化聚赖氨酸骨架上的任一个或多个末端氨基(-nh2)与二乙烯三胺五乙酸发生酰基化反应,获得偶联有二乙烯三胺五乙酸基的超支化聚赖氨酸大分子。
未反应的二乙烯三胺五乙酸通过在蒸馏水中进行透析(截留分子量为3500)除去,然后冻干透析液,获得纯净的偶联有二乙烯三胺五乙酸的所述第一超支化聚赖氨酸分子(hbpll-dtpa)。
步骤三:将六水合氯化钆溶解在水中配成氯化钆浓度不低于100mg/ml的水溶液;再取上述第一超支化聚赖氨酸分子溶解在水中,将所述氯化钆水溶液滴加到第一超支化聚赖氨酸分子的水溶液中,形成第二反应液。其中,控制所述六水合氯化钆的投料量为第一超支化聚赖氨酸分子质量的1倍以上。
同时,用1mol/l氢氧化钠水溶液调节使第二反应液ph保持在6左右,控制反应温度在42℃反应6h后,使得gd3+络合至所述二乙烯三胺五乙酸基中形成络合物基团,获得主要产物为络合有钆离子的第二超支化聚赖氨酸分子。透析(截留分子量为3500)除去未反应的氯化钆,获得透析液,冻干透析液,获得所述第二超支化聚赖氨酸分子(hbpll-dtpa-gd)。
步骤四:将叶酸-n-羟基琥珀酸亚胺活性酯(fa-nhs)溶解在二甲基亚砜(dmso)中,配成10mg/ml浓度的溶液备用。将上述所获得的第二超支化聚赖氨酸分子溶解在ph为9.6的碳酸盐缓冲液中,将fa-nhs溶液滴加至所述第二超支化聚赖氨酸分子溶液中,fa-nhs的投料量为第二超支化聚赖氨酸分子质量的0.1倍以上,形成第三反应液。第三反应液在室温下反应5h,使得所述第二超支化聚赖氨酸分子在还没有偶联有其他官能团(例如二乙烯三胺五乙酸基)的任一个或多个末端氨基(-nh2)上继续与fa-nhs发生酰基化反应,使得末端氨基上的一个h原子被叶酸基所取代,形成进一步功能化的超支化聚合物,未反应的叶酸活性酯通过超滤(截留分子量为3500)除去,获得超滤滤液,冻干所述超滤滤液,获得所述超支化聚合物(fa-hbpll-dtpa-gd,如图1所示)。这种超支化聚合物可作为造影剂(如下亦称本发明造影剂)在医学领域应用。
上述步骤一至步骤四可通过以下化学方程式表示:
下面,通过几种项目性能测试展示本发明这种超支化聚合物(fa-hbpll-dtpa-gd)作为造影剂的应用优势。
性能测试一
在0.5t的mri测试仪上测试本发明造影剂与临床使用造影剂(gd-dtpa)弛豫时间t1及t1加权成像,其操作方法包括:
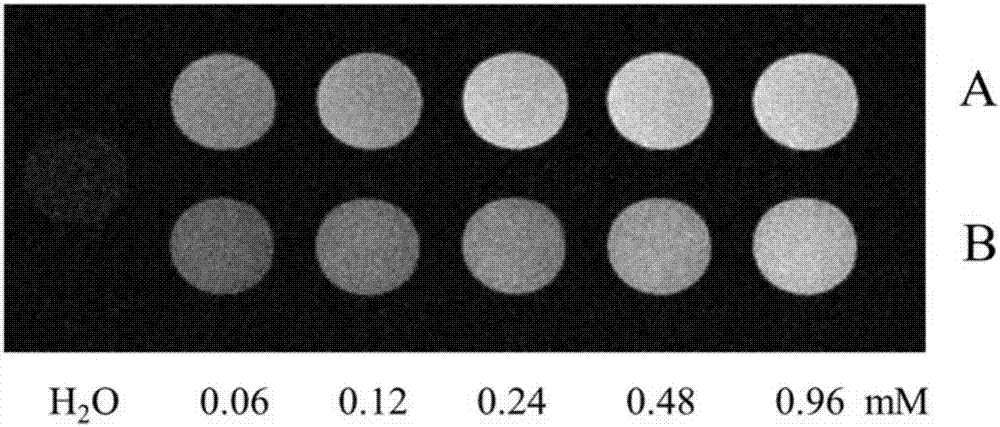
分别配制浓度为0.06~0.96mmol/l(mmol/l可简写为mm)的上述两种样品,0.5t的mri测试仪上测试后,以钆离子浓度为横坐标,纵向弛豫时间的倒数为纵坐标进行线性拟合得到本发明的造影剂和gd-dtpa的弛豫率分别为13.44mm-1·s-1和4.3mm-1·s-1(如图1所示),可见本发明的造影剂弛豫率明显高于gd-dpta。
通过两者在不同浓度下的t1加权成像(图2,a为本发明造影剂,b为gd-dpta)可以看出,随着溶液浓度的增加,两者均有变亮的趋势,但本发明造影剂的造影效果明显亮于gd-dtpa。
性能测试二
造影剂对靶细胞及正常非癌细胞毒性检测
用四唑盐比色法(wst法)来测定本发明造影剂和gd-dtpa在人口腔表皮样癌细胞(kb细胞)或293t细胞(293细胞是转染腺病毒e1a基因的人肾上皮细胞系,293t细胞由293细胞派生)中的细胞毒性。
在96孔板中以每孔5000~8000细胞的密度种入kb细胞或293t细胞悬液100μl,将96孔板置于co2培养箱中,在37℃中培养24h。将本发明造影剂或gd-dtpa溶于完全培养基中,过滤除菌;再用完全培养基(没有加入造影剂或gd-dtpa的培养基)将本发明造影剂稀释成若干组浓度为0.01~5mm不等的a组培养基;再用完全培养基将gd-dtpa稀释成若干组0.01~3.5mm不同浓度的b组培养基。
将96孔板中的旧培养基吸出,然后再将不同浓度的a组培养基或b组培养基加入到96孔板中,每孔加100μl,对照组加入100μl完全培养基,继续培养24h。最后移出所有培养基,每孔加入100μl新鲜完全培养基,然后每孔加入10μlwst-1(是一种类似于mtt的化合物,在电子耦合试剂存在的情况下,可以被线粒体内的一些脱氢酶还原生成橙黄色的甲臜(formazan)。细胞增殖越多越快,则颜色越深;细胞毒性越大,则颜色越浅。中英文全称为2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2h-tetrazolium,monosodiumsalt,2-(4-碘代苯)-3-(4-硝基苯)-5-(2,4二硫代苯)-2h-四唑盐单钠盐),置于培养箱中培养2h,用酶标仪测定450nm处的吸收值od450nm。每个造影剂或gd-dtpa浓度(称为实验组)及对照组做4个平行样。根据吸光值计算细胞的相对存活率。空白组即不加细胞的完全培养基,对照组即不加a组培养基或b组培养基的细胞。
细胞相对存活率(%)=100×(实验组od-空白组od)/(对照组od-空白组od)
如图3所示,kb细胞与fa-hbpll-dtpa-gd孵育后的存活率与浓度相关:在gd3+浓度低于2mm时,存活率均接近100%;gd3+浓度至2.5mm时存活率在90%以上。表明本发明造影剂分子浓度在2mm以下时无生物毒性,2.5mm左右有较低的生物毒性。
如图4所示,293t细胞与fa-hbpll-dtpa-gd孵育后的存活率较高,在gd3+浓度为5mm时存活率仍然在100%以上。表明本发明造影剂分子浓度在5mm以下时对正常细胞无生物毒性。
性能测试三
造影剂的组织毒理检测
用苏木精-伊红染色法(hematoxylin-eosinstaining,h&e染色法)来测定本发明造影剂在正常无胸腺裸鼠体内的组织毒性。
四周龄正常无胸腺裸鼠被分为三组:
第一组尾静脉注射生理盐水作为对照组;
第二组尾静脉注射含有本发明造影剂的生理盐水溶液,其中钆离子浓度为0.1mm/kg;
第三组尾静脉注射含有本发明造影剂的生理盐水溶液,其中钆离子浓度为0.3mm/kg。
正常条件下饲养2天后颈脱位处死,收集心、肝、脾、肺、肾进行h&e切片染色,并用显微镜拍照观察。
如图5所示,本发明造影剂对各个脏器组织的损伤较小,高浓度样本无明显提升。具体来看,肝脏切片中的肝细胞比较正常,并没有任何炎症反应的迹象。在肺部切片中也观察不到肺纤维化。所有其他的切片样本也未观察到任何的组织坏死的情况。表明,本发明所述的造影剂对重要脏器无明显的病理改变或损伤,提示这种造影剂具有非常好的生物相容性、安全性。
性能测试四
造影剂的靶向细胞摄取实验
用激光共聚焦荧光显微镜来研究造影剂分子的细胞摄取,从而证明其靶向性。
首先将fa-hbpll-dtpa-gd进行荧光标记。将kb细胞以20000每孔的密度种入含有细胞爬板的24孔板内,在培养箱中培养约24h后,细胞密度达到约60%。将细胞爬板分为3组:
第一组加入100微升磷酸缓冲盐溶液(pbs)作为对照组;
第二组加入含有本发明造影剂(钆离子浓度为2mm)的溶液;
第三组预先使用5mm叶酸培养一小时后加入本发明造影剂(钆离子浓度为2mm)的溶液作为抑制组。
继续在培养箱中培养1h后用pbs洗5遍,加入质量分数为4%的多聚甲醛溶液固定细胞,在培养箱中放置半小时后取出,用pbs洗3遍,用4',6-二脒基-2-苯基吲哚(dapi)对细胞核进行染色,在培养箱中放置半小时后取出,用pbs洗5遍,固定在玻璃板上进行激光共聚焦荧光显微镜拍照。
如图6所示,第一行中对照组没有观察到绿色荧光,第三行的荧光标记的fa-hbpll-dtpa-gd组中可以观察到很强的荧光信号,而第二行的抑制组中未观察到荧光信号。这些结果说明叶酸基修饰后的大分子造影剂的确是通过叶酸受体介导的细胞内吞,证明了其靶向性。
性能测试五
造影剂的体内mri成像实验
构建kb荷瘤裸鼠模型并分为三组:分别为对照组,非靶向组和靶向组。首先,通过腹腔注射20%的乌拉坦溶液,剂量为5ml/kg体重。待三组老鼠都进入深度麻醉后,进行未注射造影剂前的空白扫描。然后,对照组通过尾静脉注射gd-dtpa,非靶向组通过尾静脉注射hbpll-dtpa-gd,靶向组通过尾静脉注射fa-hbpll-dtpa-gd,三组的钆离子剂量都是0.1mmol/kg体重。然后将裸鼠固定好,置于1.5t的微型磁共振成像仪器内,在注射后1h,3h,5h,8h以及24h五个时间点拍摄t1加权磁共振图像。
如图7所示,在以0.1mmol/kg的注射量由尾静脉将三组造影剂注入裸鼠体内以后,三组呈现出了明显不同的成像增强效果。为了使三组mri扫描所得的图片有相互对比性,序列参数统一设置为te=14.26ms以及tr=100ms。在靶向组中,磁共振成像信号得到明显增强,图像更亮,肿瘤组织与周围组织的对比度更大。并且在每一个时间点都显示出优于非叶酸靶向组和对照组的效果,表明本发明所述的大分子造影剂具有肿瘤靶向性以及明显的成像对比增强。
以上所述仅是本申请的具体实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本申请原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!