苄基-1H-吡唑、苄基-1H-吡咯衍生物及其制备方法和用途与流程
本发明属于化学医药领域,具体涉及苄基-1H-吡唑、苄基-1H-吡咯衍生物及其制备方法和用途。
背景技术:
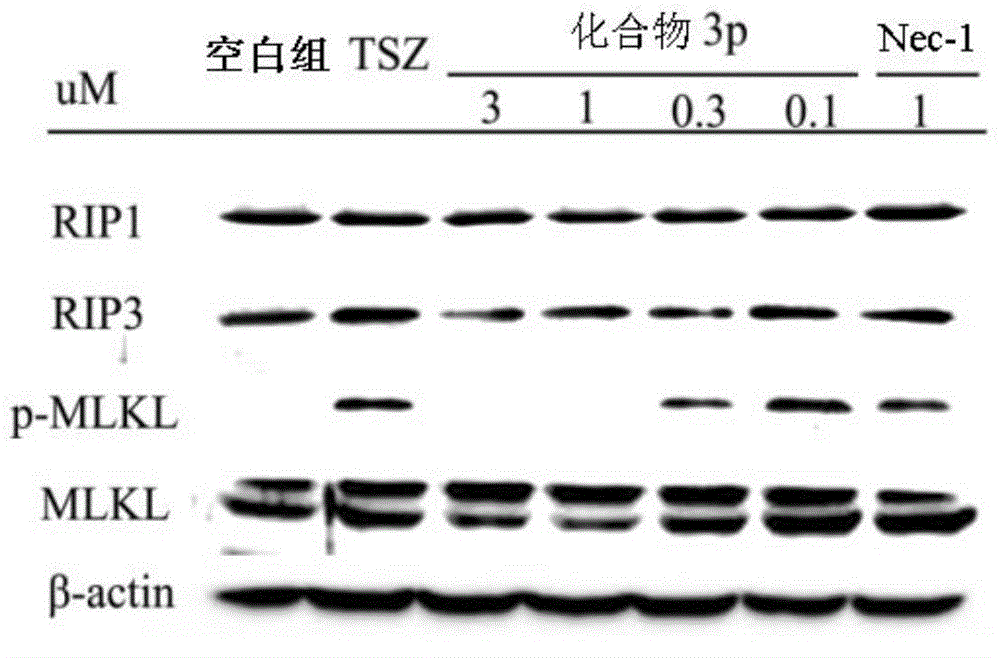
:细胞程序性死亡是近年研究发现的一种新型细胞死亡方式,是具有传统坏死样细胞死亡形态但细胞呈现程序性死亡的特殊细胞死亡方式。通常在凋亡通路被抑制的情况下发生,程序性死亡的细胞内兼具凋亡和坏死混合型特征。受体相互作用蛋白1(RIP1)是决定细胞生死的交叉点,在细胞的存活与凋亡或程序性坏死等过程中发挥重要的作用。RIP1与另一受体相互作用蛋白3(RIP3)形成的复合物是细胞发生程序性坏死的关键,而RIP1是程序性坏死过程中关键的调控因子。RIP1的第一个小分子抑制剂Nec-1在多种人类疾病动物模型中均显示了明显的作用,如缺血性损伤、神经退行性疾病、免疫相关疾病及恶性肿瘤等。选择性的抑制RIP1激酶活性也成为了治疗相关疾病的主要策略之一。以下为已报到的坏死抑制剂:上述已报到的坏死抑制剂大部分都活性较差,且药代性质不佳,这限制了其进一步的研究。因此,对RIP1激酶更佳深入的研究以期能为临床应用提供新的可行性靶点成为目前的研究难点和热点。研发更多的RIP1激酶抑制剂从中找到活性更佳体内效果更好的具有临床应用价值的小分子抑制剂具有广阔的应用前景。技术实现要素:本发明提供了一种苄基-1H-吡唑、苄基-1H-吡咯衍生物,其结构如式Ⅰ所示:其中,X为C、N或O;R1、R2、R5~R7独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子。作为本发明优选的技术方案,X为C或N;R1、R2、R5~R7独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子。优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。进一步优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N或O原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。更进一步优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个O原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。最优的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为不饱和的六元环或饱和的六元杂环;所述的六元杂环含有2个O原子;R5~R7独立地为-H、-NH2、卤素、甲基、-CN、甲氧基、或-CF3。上述的苄基-1H-吡唑、苄基-1H-吡咯衍生物,当X为N,R1为-NO2时,其结构如式Ⅱ所示:其中,R2、R5~R7独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子。优选的,R2为-H、-NO2、-COOH、-NH2或卤素;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。优选的,R2为-H或卤素;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。进一步优选的,R2为-H;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N或O原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。更进一步优选的,R2为-H;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个O原子;R5~R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、或-CF3。最优的,R2为-H;R3、R4独立地为-H、-NO2、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为不饱和的六元环或饱和的六元杂环;所述的六元杂环含有2个O原子;R5~R7独立地为-H、-NH2、卤素、甲基、-CN、甲氧基、或-CF3。上述的苄基-1H-吡唑、苄基-1H-吡咯衍生物,当R3和R6为-F时,其结构如式Ⅲ所示:其中,X为C或N;R1、R2、R5、R7独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基或-CF3;R4为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3。优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R4为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;R5、R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基或-CF3。进一步优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R4为-H;R5、R7独立地为-H、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基或-CF3。更进一步优选的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R4为-H;R5、R7独立地为-H或C1~C4烷基。最优的,X为C或N;R1、R2独立地为-H、-NO2、-COOH、-NH2或卤素;R4为-H;R5、R7独立地为-H。上述的苄基-1H-吡唑、苄基-1H-吡咯衍生物,其结构式如下所示:本发明还提供了上述苄基-1H-吡唑、苄基-1H-吡咯衍生物的制备方法。合成路线为:其中,X为C、N或O;Y为卤素;R1、R2、R5~R7独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基或-CF3;R3、R4独立地为-H、-NO2、-COOH、-NH2、卤素、C1~C4烷基、-CN、C1~C4烷氧基、-CF3;或R3和R4环合为饱和的六元环、不饱和的六元环或饱和的六元杂环;所述的六元杂环含有1~3个N、O或S原子。上述苄基-1H-吡唑、苄基-1H-吡咯衍生物的制备方法,包括以下操作步骤:(1)首先在有机溶剂中,将3-R1吡唑/吡咯与卤代试剂反应,得到3-R1-4-Br吡唑/;(2)再与反应得到式Ⅰ化合物;其中,步骤(1)中所述的有机溶剂为丙酮、N,N-二甲基甲酰胺、氯仿惰性溶剂等。所述 的卤代试剂为NBS、NIS、NCS或卤素,其用量为1~2个当量。所述反应的温度为0~90℃,反应的时间为3~12h。其中,步骤(2)中所述反应的溶剂选用丙酮、N,N-二甲基甲酰胺、氯仿等。所述反应的温度为0~100℃,反应的时间为0.5~3h。同时,所述反应中要加入1~2个当量的K2CO3和催化量的四丁基溴化铵。上述苄基-1H-吡唑、苄基-1H-吡咯衍生物包括了它们的同位素化合物、外消旋体、旋光活性异构体、多晶型形式或其混合物。本发明还提供了式Ⅰ~Ⅲ所示的苄基-1H-吡唑、苄基-1H-吡咯衍生物药学上可接受的盐或水合物。本发明还提供了上述苄基-1H-吡唑、苄基-1H-吡咯衍生物的前药,依据本发明,前药是上述化合物的衍生物,它们自身可能具有较弱的活性或甚至没有活性,但是在给药后,在生理条件下(例如通过代谢、溶剂分解或另外的方式)被转化成相应的生物活性形式。一种药物组合物,是由式Ⅰ~Ⅲ所示的苄基-1H-吡唑、苄基-1H-吡咯衍生物、及其盐或水合物添加药学上可以接受的辅助性成分制备而成的。式Ⅰ~Ⅲ所示的苄基-1H-吡唑、苄基-1H-吡咯衍生物、及其盐或水合物在制备RIP1激酶抑制剂中的用途。式Ⅰ~Ⅲ所示的苄基-1H-吡唑、苄基-1H-吡咯衍生物、及其盐或水合物在制备治疗缺血性损伤、神经退行性疾病、免疫相关疾病或恶性肿瘤药物中的用途。本发明提供的苄基-1H-吡唑、苄基-1H-吡咯衍生物,对RIP1激酶抑制活性显著,体内治疗缺血性心肌细胞坏死效果明显。该系列衍生物的溶解性较好,较Nec系列化合物在疗效和药代性质上均有较大改善。同时,本发明提供的苄基-1H-吡唑、苄基-1H-吡咯衍生物,其合成方法简单,适合大规模生产。附图说明图1化合物3p及Nec-1的细胞活性测试曲线图。图2化合物3p及Nec-1的免疫蛋白印迹实验。图3化合物3p及Nec-1的免疫共沉淀实验。图4化合物3p治疗后心肌细胞HE染色结果图。其中,a为假手术组,b为空白组,c为实验组。图5化合物4f及Nec-1的细胞活性测试曲线图。图6化合物4f的免疫蛋白印迹实验。图7化合物4f的免疫共沉淀实验。图8化合物4f治疗后胰腺组织HE染色结果图。其中,a为假手术组,b为空白组,c为实验组。具体实施方式实施例14-氯-3-硝基-1H-吡唑(中间体2a)的制备向反应瓶中加入3-硝基-1H-吡唑(5mmol)、NBS(5.5mmol),加入20mLDMF(N,N-二甲基甲酰胺),然后加热至90℃,氮气保护下反应过夜。反应完全后,减压蒸馏除去大部分DMF,加水后析出大量固体,过滤,20mL水洗,固体干燥后得目标产品,淡黄色固体。(收率87%)1H-NMR(400MHz,DMSO-d6)δ:14.32(s,1H),8.37(s,1H);MS(ESI),m/z:148.1[M+H]+。中间体2b,2c用上述相同方法制备得到。实施例24-溴-3-硝基-1H-吡唑(中间体2b)的制备采用与制备中间体2a相同方法,制备得到中间体2b,产率55%。1H-NMR(400MHz,DMSO-d6)δ:14.38(s,1H),8.45(s,1H);m/z:192.2[M+H]+。实施例34-碘-3-硝基-1H-吡唑(中间体2c)的制备采用与制备中间体2a相同方法,制备得到中间体2c,产率20%。1H-NMR(400MHz,DMSO-d6)δ:14.31(s,1H),8.27(s,1H);MS(ESI),m/z:148.1[M+H]+。实施例44-溴-1-(2,5-二氟苄基)-3-硝基-1H-吡唑(化合物4f)的制备向50mL反应瓶中加入3-硝基-4-溴-1H-吡唑(8.9mmol)、2,5-二氟溴苄(9.7mmol)、碳酸钾(26.7mmol)和催化量四丁基溴化铵,加入20mLN,N-二甲基甲酰胺,常温下搅拌1h。反应完全后将反应液10mL水中,析出大量白色固体,过滤,将固体干燥后得到黄色固体即 为产品。(收率92%)1H-NMR(400MHz,DMSO-d6)δ:8.50(s,1H),7.33(dd,J=12.4,8.0Hz,3H),5.52(s,2H);MS(ESI),m/z:274.2[M+H]+。实施例51-(萘-1-基甲基)-3-硝基-1H-吡唑(化合物3a)的制备以1-溴甲基萘和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3a,产率85%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.18(s,1H),8.17(d,J=4.4Hz,1H),7.98(t,J=9.6Hz,2H),7.63-7.52(m,3H),7.43(d,J=7.2Hz,1H),7.08(d,J=2.4Hz,1H),5.99(s,2H);MS(ESI),m/z:254.1[M+H]+。实施例61-((2,3-二氢苯并[b][1,4]二氧六环-6-基)甲基)-3-硝基-1H-吡唑(化合物3b)的制备以1-溴甲基苯并二氧六环和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3b,产率:75%,白色固体。1HNMR(400MHz,DMSO-d6)δ:8.12(d,J=2.5Hz,1H),7.05(d,J=2.5Hz,1H),6.89(d,J=1.4Hz,1H),6.88–6.78(m,2H),5.32(s,2H),4.23(s,4H);MS(ESI),m/z:261.9[M+H]+。实施例74'-((3-硝基-1H-吡唑-1-基)甲基)-[1,1联苯]-2-氰基(化合物3c)的制备以4'-溴甲基-[1,1联苯]-2-氰基和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3c,产率:97%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.25(d,J=2.8Hz,1H),7.96(d,J=7.2Hz,1H),7.80(t,J=7.6Hz,1H),7.63(d,J=6.8Hz,2H),7.59(d,J=8.4Hz,2H),7.49(d,J=8.4Hz,2H),7.12(d,J=2.4z,1H),5.58(s,2H);MS(ESI),m/z:305.4[M+H]+。实施例81-苄基-3-硝基-1H-吡唑(化合物3d)的制备以苄基和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3d,产率:90%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.17(d,J=2.8Hz,1H),7.41-7.33(m,5H),7.08(d, J=2.8Hz,1H),5.48(s,2H);MS(ESI),m/z:204.2[M+H]+。实施例91-(2-氯苄基)-3-硝基-1H-吡唑(化合物3e)的制备以2-氯氯苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3e,产率:87%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.15(d,J=2.4Hz,1H),7.53(dd,J=8.0,2.0Hz,1H),7.40(qd,J=7.2,2.0Hz,2H),7.26(dd,J=7.2,2.0Hz,1H),7.10(d,J=2.8Hz,1H),5.60(s,2H);MS(ESI),m/z:238.1[M+H]+。实施例101-(3-氯苄基)-3-硝基-1H-吡唑(化合物3f)的制备以3-氯氯苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3f,产率:85%,白色固体。1HNMR(400MHz,DMSO-d6)δ:8.19(s,1H),7.43(d,J=4.0Hz,3H),7.29(s,1H),7.09(s,1H),5.50(s,2H);MS(ESI),m/z:238.3[M+H]+。实施例111-(4-氯苄基)-3-硝基-1H-吡唑(化合物3g)的制备以4-氯氯苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3g,产率:86%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.18(d,J=2.8Hz,1H),7.46(d,J=8.4Hz,2H),7.36(d,J=8.4Hz,2H),7.09(d,J=2.4Hz,1H),5.48(s,2H);MS(ESI),m/z:238.3[M+H]+。实施例121-(4-甲氧基苄基)-3-硝基-1H-吡唑(化合物3h)的制备以4-甲氧基溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3h,产率:89%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.12(d,J=2.4Hz,1H),7.32(d,J=8.8Hz,2H),7.05(d,J=2.8Hz,1H),6.94(d,J=8.8Hz,2H),5.38(s,2H),3.75(s,3H);MS(ESI),m/z:234.2[M+H]+。实施例134-((3-硝基-1H-吡唑-1-基)甲基)苯胺(化合物3i)的制备以4-氨基溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3i,产率:45%,黄色固体。1H-NMR(400MHz,DMSO-d6)δ:8.03(d,J=2.4Hz,1H),7.05(d,J=8.0Hz,2H),7.03(d,J=2.4Hz,1H),6.54(d,J=8.4Hz,2H),5.22(s,2H),3.18(s,2H);MS(ESI),m/z:219.1[M+H]+。实施例143-硝基-1-(4-(三氟甲基)苄基)-1H-吡唑(化合物3j)的制备以4-三氟甲基溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3j,产率:92%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.22(d,J=2.4Hz,1H),7.77(d,J=8.0Hz,2H),7.53(d,J=8.0Hz,2H),7.12(d,J=2.4Hz,1H),5.61(s,2H);MS(ESI),m/z:272.1[M+H]+。实施例151-(4-溴苄基)-3-硝基-1H-吡唑(化合物3k)的制备以4-溴溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3k,产率:93%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.17(d,J=2.4Hz,1H),7.60(d,J=8.4Hz,2H),7.30(d,J=8.4Hz,2H),7.09(d,J=2.4Hz,1H),5.47(s,2H);MS(ESI),m/z:281.9[M+H]+。实施例161-(2-甲基苄基)-3-硝基-1H-吡唑(化合物3l)的制备以2-甲基溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3l,产率:96%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.08(d,J=2.4Hz,1H),7.26-7.18(m,3H),7.11(s,1H),7.08(d,J=2.4Hz,1H),5.50(s,2H),2.31(s,3H);MS(ESI),m/z:218.1[M+H]+。实施例171-(2-溴苄基)-3-硝基-1H-吡唑(化合物3m)的制备以2-溴溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3m,产率:92%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.14(d,J=2.4Hz,1H),7.70(d,J=7.6Hz,1H),7.42(t,J=7.6Hz,1H),7.33(t,J=7.6Hz,1H),7.19(d,J=7.6Hz,1H),7.11(d,J=2.8Hz,1H),5.57(s,2H);MS(ESI),m/z:282.1[M+H]+。实施例181-(3,4-二氟苄基)-3-硝基-1H-吡唑(化合物3n)的制备以3,4-二氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3n,产率:90%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.18(d,J=2.8Hz,1H),7.50-7.43(m,2H),7.23-7.20(m,1H),7.09(d,J=2.4Hz,1H),5.48(s,2H);MS(ESI),m/z:240.5[M+H]+。实施例191-(2,4-二氟苄基)-3-硝基-1H-吡唑(化合物3o)的制备以2,4-二氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3o,产率:91%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.15(d,J=2.4Hz,1H),7.49(q,J=8.4Hz,1H),7.33(td,J=8.4,2.4Hz,1H),7.15(td,J=8.4,2.4Hz,1H),7.08(d,J=2.4Hz,1H),5.52(s,2H);MS(ESI),m/z:240.6[M+H]+。实施例201-(2,5-二氟苄基)-3-硝基-1H-吡唑(化合物3p)的制备以2,5-二氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3p,产率:98%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.17(d,J=2.4Hz,1H),7.34-7.24(m,3H),7.09(d,J=2.4Hz,1H),5.54(s,2H);MS(ESI),m/z:240.1[M+H]+。实施例211-(3,5-二氟苄基)-3-硝基-1H-吡唑(化合物3q)的制备以3,5-二氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3q,产率:94%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.20(d,J=2.4Hz,1H),7.24(t,J=9.2Hz,1H),7.09(d,J=8.4Hz,2H),7.07(d,J=2.4Hz,1H),5.52(s,2H);MS(ESI),m/z:240.2[M+H]+。实施例221-(2-氯-4-氟苄基)-3-硝基-1H-吡唑(化合物3r)的制备以2-氯-4-氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3r,产率:95%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.15(d,J=2.8Hz,1H),7.50(dd,J=8.8,2.4Hz,1H),7.40(t,J=7.2Hz,1H),7.10(td,J=8.4,2.4Hz,1H),7.10(d,J=2.8Hz,1H),5.57(s,2H);MS(ESI),m/z:256.0[M+H]+。实施例231-(2,5-二氯苄基)-3-硝基-1H-吡唑(化合物3s)的制备以2,5-二氯氯苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3s,产率:90%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.19(d,J=2.4Hz,1H),7.57(d,J=8.4Hz,1H),7.51(dd,J=8.8,2.4Hz,1H),7.42(d,J=2.0Hz,1H),7.11(d,J=2.4Hz,1H),5.58(s,2H);MS(ESI),m/z:272.1[M+H]+。实施例241-(5-氯-2-氟苄基)-3-硝基-1H-吡唑(化合物3t)的制备以5-氯-2-氟溴苄和3-硝基-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物3t,产率:97%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:8.18(d,J=2.4Hz,1H),7.53-.7.49(m,2H),7.36-7.32(m,1H),7.09(d,J=2.4Hz,1H),5.54(s,2H);MS(ESI),m/z:256.2[M+H]+。实施例251-(3-氯-4-((3-硝基-1H-吡唑-1-基)甲基)苯基)-4-甲基哌啶(化合物3u)的制备将化合物3r(100mg,0.4mmol)置于100mL圆底烧瓶中,加入3mLN-甲基哌啶,滴 加三乙胺(155mg,1.2mmol),常温反应过夜。反应完全后,加入10mL水中,析出大量固体,过滤,减压干燥得到化合物3u,黄色固体,产率75%。1H-NMR(400MHz,DMSO-d6)δ:8.03(d,J=2.4Hz,1H),7.25(d,J=8.8Hz,1H),7.05(d,J=2.4Hz,1H),7.00(d,J=2.4Hz,1H),6.94(d,J=2.0Hz,1H),6.92(d,J=2.4Hz,1H),5.43(s,2H),3.19(t,J=4.4Hz,4H),2.42(d,J=4.4Hz,4H),2.21(s,3H);MS(ESI),m/z:336.1[M+H]+。实施例261-(2,5-二氟苄基)-1H-吡唑(化合物4a)的制备以2,5-二氟溴苄和吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4a,产率:87%,蓝色固体。1H-NMR(400MHz,DMSO-d6)δ:7.88(d,J=2.1Hz,1H),7.52(d,J=1.4Hz,1H),7.28(dt,J=9.2,4.6Hz,1H),7.21(dd,J=8.0,3.8Hz,1H),6.94(ddd,J=8.9,5.7,3.2Hz,1H),6.31(t,J=1.8Hz,1H),5.42(s,2H);MS(ESI),m/z:195.2[M+H]+。实施例271-(2,5-二氟苄基)-1H-吡唑-3-羧酸(化合物4b)的制备以2,5-二氟溴苄和3-羧基吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4b,产率:75%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:12.75(s,1H),7.95(d,J=2.0Hz,1H),7.35–7.14(m,3H),7.08(d,J=8.4Hz,1H),5.51(s,2H);MS(ESI),m/z:237.1[M-H]+。实施例281-(2,5-二氟苄基)-1H-吡唑-3-胺(化合物4c)的制备以2,5-二氟溴苄和3-氨基吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4c,产率:75%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:7.12(t,J=5.3Hz,1H),7.03–6.93(m,1H),6.90–6.79(m,1H),6.79–6.67(m,1H),5.46(d,J=1.2Hz,1H),5.45(s,2H),4.24(s,2H);MS(ESI),m/z:210.1[M+H]+。实施例291-(2,5-二氟苄基)-4-硝基-1H-吡唑(化合物4d)的制备以2,5-二氟溴苄和4-硝基吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4d,产率:75%,白色固体。1H-NMR(400MHz,DMSO-d6)δ:9.05(s,1H),8.30(s,1H),7.30(dtd,J=16.2,8.6,5.1Hz,3H),5.47(s,2H);MS(ESI),m/z:240.2[M+H]+。实施例304-氯-1-(2,5-二氟苄基)-3-硝基-1H-吡唑(化合物4e)的制备以2,5-二氟溴苄和3-硝基-4-氯-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4e,产率:92%,黄色固体。1H-NMR(400MHz,DMSO-d6)δ:8.50(s,1H),7.33(dd,J=12.4,8.0Hz,3H),5.52(s,2H);MS(ESI),m/z:274.2[M+H]+。实施例314-溴-1-(2,5-二氟苄基)-3-硝基-1H-吡唑(4f)的制备以2,5-二氟溴苄和3-硝基-4-溴-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4f,产率:89%,棕色固体。1H-NMR(400MHz,DMSO-d6)δ:8.49(s,1H),7.48–7.20(m,3H),5.53(s,2H);MS(ESI),m/z:318.2[M+H]+。实施例321-(2,5-二氟苄基)-4-碘-3-硝基-1H-吡唑(化合物4g)的制备以2,5-二氟溴苄和3-硝基-4-碘-1H-吡唑为原料,采用与实施例4相同的操作步骤,制备得到化合物4g,产率:90%,黄色固体。1H-NMR(400MHz,DMSO-d6)δ:8.37(s,1H),7.54–7.11(m,3H),5.52(s,2H);MS(ESI),m/z:365.9[M+H]+。实施例331-(2,5-二氟苄基)-4-碘-3-硝基-1H-吡咯(化合物4h)的制备以2,5-二氟溴苄和3-硝基-1H-吡咯为原料,采用与实施例4相同的操作步骤,制备得到化合物4h。1HNMR(400MHz,DMSO)δ8.15(t,J=1.9Hz,1H),7.23(tt,J=9.4,2.2Hz,1H),7.17–7.09(m,2H),7.07(t,J=2.7Hz,1H),6.71(dd,J=2.9,1.9Hz,1H),5.22(s,2H);MS(ESI),m/z:239.1[M+H]+。实施例34细胞程序性坏死抑制活性实验1、细胞株:HT-29细胞购置于美国ATCC(美国菌种保藏中心),并保种于四川大学生物治疗国家重点实验室。2、细胞水平测试试验方法:选用HT-29细胞株,按常规细胞培养法进行培养。采用四氮唑盐MTT还原法,将药物与细胞共培养24小时,贴壁细胞需先贴壁24小时后再给药。给药同时加肿瘤坏死因子TNFα(购于Peprotech公司),凋亡抑制蛋白cIAP的抑制剂Smacmimetic(购于Selleck公司),含半胱氨酸的天冬氨酸水解酶caspase的抑制剂Z-VAD-FMK(购于APExBIO公司)以诱导细胞产生坏死。同时设阳性对照和诱导组和空白组,考察化合物3p抑制细胞程序性坏死的作用。具体操作如下:a)选用对数生长期的贴壁HT-29细胞,用0.25%胰酶(购于Hyclone公司,SH30042.01)消化后,用含10%胎牛血清(购自兰州民海生物公司)的相应的DMEM完全培养基(购于Gibco公司,11965-92)配成4000个/mL的细胞悬液,接种在96孔培养板中,每孔接种200μL,37℃,5%CO2培养24h。实验组换新的含不同浓度被测样品的培养基,对照组则换含等体积溶剂的培养基,实验组和对照组同时加入TNFα,Smacmimetic,Z-VAD-FMK以诱导,空白组不加,每组设3~5个平行孔,37℃,5%CO2培养1d。b)弃去上清液,每孔加入200μL新鲜配制的含0.2mg/mLMTT(购于Thermoscientific公司)的无血清培养基.37℃继续培养4h。小心弃上清,并加入200μLDMSO(二甲基亚砜),用微型超声振荡器混匀后,在酶标仪上以试验波长为570nm,参比波长为450nm测定光密度值。c)按下式计算药物对肿瘤细胞生长的抑制率:细胞程序性坏死抑制率%=(OD实验-OD诱导/OD空白)×100%。以同一样品的不同浓度对细胞程序性坏死抑制率作图可得到剂量反应曲线,从中求出样品的半数有效浓度EC50。表1中的Nec-1购于SELLECK公司,纯度≥98%,其结构式为:表1列举了本发明化合物与Nec-1对细胞程序性坏死的半数有效浓度EC50。表1本发明化合物与Nec-1对细胞程序性坏死的半数有效浓度EC50化合物EC50(uM)化合物EC50(uM)3a>103p0.2923b>103q1.8723c>103r0.8273d2.4603s53e2.5393t33f33u>103g1.4464a>103h54b0.4183i54c>103j5-104d>103k1.9614e0.1833l1.1564f0.1573m0.554g0.4113n34h>103o0.876Nec-11.051从表1中可以看出,本发明提供的化合物大部分都表现出了良好的抗细胞程序性坏死活性,其中化合物3m、3o、3p、3r、4b、4e、4f和4g具有<1uM的抑制活性。实施例35体外活性实验1、细胞株:HT-29细胞购置于美国ATCC(美国菌种保藏中心),并保种于四川大学生物治疗国家重点实验室。2、细胞水平测试试验方法:选用HT-29细胞株,按常规细胞培养法进行培养。采用四氮唑盐MTT还原法,将药物与细胞共培养24小时,贴壁细胞需先贴壁24小时后再给药。给药同时加肿瘤坏死因子TNFα (购于Peprotech公司),凋亡抑制蛋白cIAP的抑制剂Smacmimetic(购于Selleck公司),含半胱氨酸的天冬氨酸水解酶caspase的抑制剂Z-VAD-FMK(购于APExBIO公司)以诱导细胞产生坏死。同时设阳性对照和诱导组和空白组,考察化合物3p抑制细胞程序性坏死的作用。具体操作如下:a)选用对数生长期的贴壁HT-29细胞,用0.25%胰酶(购于Hyclone公司,SH30042.01)消化后,用含10%胎牛血清(购自兰州民海生物公司)的相应的DMEM完全培养基(购于Gibco公司,11965-92)配成4000个/mL的细胞悬液,接种在96孔培养板中,每孔接种200μL,37℃,5%CO2培养24h。实验组换新的含不同浓度被测样品的培养基,对照组则换含等体积溶剂的培养基,实验组和对照组同时加入TNFα,Smacmimetic,Z-VAD-FMK以诱导,空白组不加,每组设3~5个平行孔,37℃,5%CO2培养1d。实验组即化合物3p,纯度≥98%;对照组Nec-1购于SELLECK公司,纯度≥98%。b)弃去上清液,每孔加入200μL新鲜配制的含0.2mg/mLMTT(购于Thermoscientific公司)的无血清培养基.37℃继续培养4h。小心弃上清,并加入200μLDMSO(二甲基亚砜),用微型超声振荡器混匀后,在酶标仪上以试验波长为570nm,参比波长为450nm测定光密度值。c)按下式计算药物对肿瘤细胞生长的抑制率:细胞程序性坏死抑制率%=(OD实验-OD诱导/OD空白)×100%。并按抑制率作图。化合物3p和4f对TSZ(TNFα,Smacmimetic和Z-VAD-FMK处理后的坏死诱导组)诱导HT-29产生的程序性坏死过程造成的细胞死亡抑制曲线见图1和图5。从图可以看出,化合物3p和4f对程序性坏死具有抑制作用,且抑制活性高于阳性对照Nec-1。3、蛋白质免疫印迹实验,具体操作如下:a)制备蛋白样品:培养HT-29细胞,在细胞处于对数生长期时铺于六孔板,24h后按一定浓度梯度加药,设立空白组,阴性对照组,阳性对照组(Nec-1)和实验组(化合物3p)。阴性对照组和阳性对照组以及实验组用TNFα,Smacmimetic和Z-VAD-FMK诱导necroptosis8h,后用加入苯甲基磺酰氟(PMSF,购于Sigma-Aldrich公司)、蛋白酶抑制剂Cocktail(购于Sigma-Aldrich公司)和强细胞裂解液RIPA(购于碧云天公司)裂解细胞,并用细胞超声破碎仪进行破碎,13000rpm离心15min后收集上清,用BCA定量试剂盒(购于碧云天公司)进行定量。加5X上样缓冲液(购于碧云天公司)100℃煮样10min后分装。由于p-RIP1和p-RIP3抗体未能购得,故采用免疫沉淀的方法,用磷酸化丝氨酸(Phosphoserine/ps)抗体(购于Lifetechnologies公司)获取磷酸化的相互作用蛋白激酶1即p-RIP1和磷酸化的相互作用蛋白激酶3即p-RIP3蛋白。Phosphoserine/ps抗体与定量后的蛋白进行孵育过夜后,再用蛋白 A琼脂糖珠(购于Roche公司)捕获抗原抗体复合物4h,4000rpm/2min/次离心3次收集复合物,加5Xloadingbuffer100℃煮样10min后离心,取上清电泳。用RIP1抗体(购于BDBiosciences)和RIP3抗体(购于ABcam公司)来检测p-RIP1和p-RIP3的水平。b)上样电泳:将蛋白样品加入上样槽,进行聚丙烯酰胺凝胶电泳。通常浓缩胶使用80V,分离胶使用100~120V。其中一个泳道加入1~2uL预染的分子量指示剂,作为蛋白分子量的定量标准以及初步判断转膜是否成功的标准。当指示剂溴酚蓝到达电泳槽底部时停止电泳,根据蛋白分子量标准切割目标条带。c)转膜:选用聚偏二氟乙烯膜(PVDF膜,购于Millipore公司),使用之前用甲醇浸泡5~10s。大于20kD的蛋白用0.45um的膜,小于20kD的蛋白用0.2um的膜。依据胶的大小裁剪合适的大小的膜和滤纸。将PVDF膜用纯甲醇浸泡5~10s后放入转移缓冲液中,待用。装配―三明治‖转膜匣:即海绵→4层滤纸→胶→膜→4层滤纸→海绵,每层放好后要小心赶走气泡。注意要将胶放于负极面即黑色面。然后将转移槽置于冰浴中,放入―三明治‖,倒入转移缓冲液和小冰袋,插入电极,通常使用100V转膜0.5~2h。d)免疫杂交与显色:将膜置于封闭缓冲液(含5%伊利脱脂奶粉的TBS/T(三乙醇胺缓冲盐/吐温水)溶液)中封闭1~2h,室,缓慢摇动。加入合适稀释度的一抗(通常为1:1000,含5%脱脂奶粉的TBS/T稀释),杂交袋孵育4℃过夜。TBS/T洗3次,每次10min。加入合适稀释度的二抗(通常为1:5000,含5%脱脂奶粉的TBS/T稀释),杂交袋孵育,37℃恒温孵箱中缓慢摇动孵育1—1.5h。TBS/T洗3次,每次20min。TBS洗1次,5min。泡在TBS中,待显影。发光反应时将显影液两种溶液按1︰1混合,先将PVDF膜在PE手套上稍微蘸干TBS溶液,然后迅速将显影液均匀涂抹于膜正面,用塑料薄膜覆盖膜,放入暗盒中,放入胶片曝光,洗片。根据不同条带强弱可调整曝光时间。实验结果见图2。化合物3p和4f对TSZ诱导HT29后关键蛋白相互作用蛋白激酶1即RIP1,相互作用蛋白激酶3即RIP3,MLKL(混合谱系激酶结构域样蛋白)的表达水平和磷酸化水平见图2、图3、图6和图7。可见化合物3p和4f明显抑制了TSZ诱导后RIP1的磷酸化,进一步影响了RIP1下游的RIP3和混合系列蛋白激酶样结构域MLKL的磷酸化。实施例36体内药效学实验1、动物来源:小鼠(Balb/C),8-10周龄,购自四川大学实验动物中心;雄性,饲养地点:四川大学生物治疗国家重点实验室SPF动物房(1号房)(实验动物使用许可证号:SYXK(川)2014-178)。2、心肌缺血模型构建及药物治疗:将小鼠用4%水合氯醛(购于百灵威公司)按10mL/kg的剂量麻醉后,仰卧位固定,在 气管表面剪开一小切口后将小动物呼吸机所连导管(留置针头套)插入小鼠气管,启动呼吸机,参数为呼吸频率110,呼吸比3︰1,潮气量1mL。观察到小鼠胸廓频率与呼吸机频率一致后,开胸,钝性分离,在体视镜辅助下找到左心耳,并在左心耳下方2~3mm用7-0带针尼龙线进行结扎。假手术组只开胸但并不进行左冠状动脉前降支结扎。空白组和实验组在左冠状动脉前降支结扎后,待小鼠恢复。手术2天后,实验组开始按10mg/kg/d的剂量给药化合物3p,给药10天后处死小鼠,并取心脏做HE染色。给药方案:化合物3p按10mg/kg/d口服给药10天,假手术组和空白组给对应空白溶剂。实验结果:化合物3p和对小鼠心肌缺血模型的保护作用见图4。化合物3p在按10mg/kg/d的剂量给药10天后对小鼠由于冠状动脉结扎而造成的心肌缺血模型具有保护作用,坏死区域减小。3.急性胰腺炎模型构建及药物治疗:将小鼠禁食12小时过夜后,腹腔注射8%L-精氨酸(购于百灵威公司),注射剂量为50ul/g,注射两次,中间间隔1小时。注射后实验组按10mg/kg/d的剂量给药化合物4f,给药5天后处死老鼠,取胰腺做HE染色。实验结果:化合物4f对小鼠胰腺炎模型的保护作用见图8,化合物4f在10mg/kg/d的剂量给药5天后对L-精氨酸造成的小鼠急性胰腺炎具有保护作用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种4‑氨基安替比林产品的合成工艺及其试液的配备方法与制造工艺
- 一种吡唑酮生产工艺的制造方法与工艺
- 一类含呋喃的苯基联吡唑甲酰胺类衍生物及其制备方法和用图
- N’?5?(取代苯基)?2?呋喃甲酰基?5?丙基?1h?吡唑?3?甲酸乙酯类化合物及其制备方法和应用
- 吡唑啉酮化合物及其用图
- 5-(4-氟苯基)-N–吗啉基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含醚性氧原子的全氟烷基取代吡唑环化合物及其制造方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含有色酮结构的吡唑类n‐对硝基苯基马来酰亚胺衍生物及其制备方法与应用