用于UV固化材料的酰基肟酯类化合物及其合成方法及应用与流程
本发明涉及一种光引发剂,尤其涉及一种用于UV固化材料的光敏引发剂。
背景技术:
紫外光(UV)固化,简称光固化技术,是一种高效、环保、节能、优质的材料表面处理技术,广泛应用于广告、印刷、高档商品包装、装潢、电子及通信等领域,目前光固化产品主要以UV涂料、UV油墨、UV胶黏剂、感光性印刷版材、光刻胶、光快速成型材料等形式出现。
光固化是利用紫外光照射具有化学活性的液态材料,引发其快速聚合交联,并使其瞬间固化的过程。
光固化与热固化等其他固化方式相比,有以下优势:1.速率快,光固化产品一般在几秒内即可固化,是目前各种油墨、涂料及胶黏剂中固化速度最快的;2.应用范围广泛,光固化产品可适用于多种基材,特别适用于一些热敏感性的材质,如纸张、电子元器件及塑料等:3.能耗低,光固化产品是常温快速固化,其能耗只有热固化的1/10到1/5;4.低污染,光固化产品基本不含挥发性有机物(VOC),是一类环境友好型的产品。
在日常生活中,光固化产品无处不在,并改变着我们的生活。如在板材涂装保护及装饰行业,UV木器涂料涂装可以提高板材耐磨、抗划伤和耐抗性,通过UV立体涂装还可大大提高装饰效果,主要应用在家具、实木地板的保护及装饰上;在印刷行业,UV胶印油墨的使用一改过去印刷品印油墨不干而需喷粉的弊病,并且色彩鲜艳饱和,清晰度更佳,UV油墨已成为户外大型广告和指示牌的生力军,也是高档烟酒、保健品、化妆品和食品包装的重要印刷材料;在光电子、信息和通信工业中,光刻胶是较早应用的光固化产品,特别适用于一些微电子产品的制作。如制作大规模集成电路用的远紫外光刻胶,在液晶显示器、等离子体显示器、有机电致发光显示器中的一些关键部件的制作中也都离不开光刻胶。
1968年Bayer公司开发了第一代紫外光固化木器涂料,首先实现了光固化技术的产业化。随后光固化技术迅猛发展,应用领域不断扩大,形成了一个新的产业。上世纪70、80年代,欧美辐射固化协会成立,推动了光固化技术的研究和发展,在北美、欧洲和日本等发达国家和地区,巴斯夫、拜耳、陶氏等跨国公司纷纷加盟光固化生产,目前已成为了具有一定市场规模的产业。我国从上世纪80年代开始发展光固化技术,由于原料和设备的限制,发展缓慢。进入90年代,紫外光固化技术和设备的引进大大推动了我国光固化产业的发展,进入21世纪,我国光固化产业获得了更加快速的发展,特别是光引发剂已成为世界上最大的生产和出口国,已初步形成一个新的高新产业。如今大力提倡可持续发展,建立和谐社会,并加大了环境的保护,这为我国光固化产业的发展提供了机遇。
主要由不饱和树脂及其单体材料组成的光固化材料(光固化涂料、油墨、光刻胶、RGB和BM),要使其能在紫外光、X射线或激光照射下发生聚合固化反应,必须光引发剂或增感剂。这些添加的光引发剂或增感剂能够在一定波长的紫外光、X射线或激光照射下,产生活性基团,激发光固化材料中的不饱和基团繁盛聚合反应,引起光固化材料的固化。
在光固化材料中,广泛应用的一些传统引发剂有:安息香衍生物、联苯酰缩酮类、α,α-二烷氧基苯乙酮类、α–羟基烷基苯酮类、α-氨基烷基苯酮类、酰基氧化膦类、二苯甲酮/胺类、米氏酮、噻吨酮/胺类、胺促进剂、芳香重氮盐、芳基腆鎓盐和硫鎓盐、二茂铁和二茂铁类、六芳基二咪唑类、三氮嗪类及传统肟酯类等。由于这些传统的光引发剂或多或少的存在着感光度低(聚合速率和转化率低)、溶解性差(透明度低和光刻残渣多)、氧气对光固化影响大及贮存稳定性差等缺点,因此它们及感光材料的使用受到了很大的限制,也极大的影响了感光材料的性能,特别是不能满足新一代大屏幕LCD关键部件BM和CF的制作要求。新型肟酯类光引发剂的出现,很大程度上解决了上述问题。肟酯类化合物的光化学特性最早出现在文献A.Wemer and A.Piguet,Ber.Dtsch.Chem.Ges.1904,37,4295中;而作为光引发剂的应用,最早则是出现在文献G.A.Delzenne,u.Laridon and H.Peeters,EuropeanPolymer Journal,1970,6,933-943中,商品名为DE-OS 179508和Agfa-Gevaert AG;曾较广泛商业应用的肟酯类光引发剂产品是Quantacure PDO。
这些传统的肟酯类光引发剂虽然光引发洁性高,但由于热稳定性差,而逐步被工业应用所淘汰;肟酯类光引发剂的"复活"最早出现在文献R.Malliviaet al,J.Photochem.Photobiol.A:Chemistry 2001,138,193和文献L.Lavalée et aI,J.Photochem.Photobiol.A:Chemistry 2002.151,27中,由于在肟酯化合物中引入了二苯硫醚或咔唑基团,这些基团中有较大的共轭体系和较强的分子内电子转移特性,因此极大的提高了这类肟酯化合物的稳定性和感光活性,目前广泛应用的两个代表性的肟酯类光引发剂是OXE-1和OXE-2,结构式如下所示:
其中OXE-l就是最典型的酮肟酯类光引发剂,它们主要应用于制造大屏幕LCD显示器的BM和RGB,价格昂贵,并且其结构式己被国外公司申请保护,专利公开号CN99108598和CN02811675,上述公布的结构式的合成方法繁琐,合成成本高,本结构的产品的应用性能不够好,热稳定性较差的问题。随后,涌现出了诸多具有优异光固化应用性能的酮肟酯类化合物的报道,如CN101565472A公开了一种含有环烷基的酮肟酯光引发剂,其具有很好的稳定性和溶解性。但是伴随着实践应用,此类产品被证实仍然存在应用性能不足的问题,如使用中存在安全隐患(分解产生苯类高毒性物质)、感光活性和热稳定性等常规性能需进一步提高。
现在入们对于光引发剂的效果及其分解产物的毒性、气味和迁移性等特性要求越来越高,开发具有良好的溶解性、低气味或无气味和低迁移性的良好特性的大分子光引发剂将成为未来发展的主要方向。但目前已商业化的大分子光引发剂大多价格昂贵或产品性能有一定缺陷,因此迫切需要价格低廉且性能好的产品来替代。
技术实现要素:
发明目的:本发明针对现有酰基肟酯类光引发剂应用性能的不足,本发明的目的是提供一种溶解性好、热稳定性好、反应活性高、生产成本低、价格低廉、基本无气味,低迁移性,且使用安全性高(毒性低)的酰基肟酯类化合物。
本发明的另一目的是提供所述酰基肟酯类化合物的制备方法。
本发明的再一目的是提供所述酰基肟酯类化合物在UV光固化材料中的应用。
技术方案:为了达到上述发明目的,本发明提供了一种酰基肟酯类化合物,所述酰基肟酯类光化合物具有通式Ⅰ所示结构:
所述R1和R4相同或不同,各自独立地选自
其中,所述R3选自-H、-NO2、1-8个碳原子的烷基或烷氧基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基、
所述Y1、Y2及Y3相同或不同,各自独立地选自-H、-CH3、2-8个碳原子的烷基或烷氧基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基;
所述X‘和Y’相同或不同,各自独立地选自-S-、-S-S-、-O-、-CO-;
上述R1和R4选择的取代基中,所述环状结构中一个或多个-H可以被-F、-Cl、-Br、-I、-NO2、-COOR′1、-CONR′2、-OR′3、1-8个碳原子的烷基或烷氧基或2-8个碳原子的烯烃基取代,其中R’1和R’2各自独立地选自-H、1-8个碳原子的烷基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基,R’3选自-H、1-8个碳原子的烷基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基或1-8个碳原子的碳链羰基,其中,1-8个碳原子的碳链羰基中羰基在端位,位于连接键处;
所述R2选自-H、1-20个碳原子的烷基或烷氧基、3-8个碳原子的环烷基或2-20个碳原子的烯烃基;
或者R2与R1所示基团相同,
前提是:所述R1和R4中至少一个表示
在本发明的一些优选实施方式中,所述R1和R4中至少一个表示
在本发明的一些优选实施方式中,所述R1和R4中所述环状结构中一个或多个-H可以被-F、-NO2、-COOR′1、-CONR′2、-OR′3、1-8个碳原子的烷基或烷氧基,其中R’1和R’2各自独立地选自-H、1-8个碳原子的烷基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基,R’3选自-H、1-8个碳原子的烷基或1-8个碳原子的碳链羰基,其中,1-8个碳原子的碳链羰基中羰基在端位,位于连接键处。
在本发明的一些优选实施方式中,优选地,所述R1和R4相同或不同,各自独立地选自
本发明的一些优选实施方式中,所述R2选自-H、1-20个碳原子的烷基或烷氧基、2-20个碳原子的烯烃基或与R1所示基团相同。
本发明的一些实施方式中,优选地,所述R2选自-H、1-20个碳原子的烷基或烷氧基、2-20个碳原子的烯烃基、
本发明的一些实施方式中,优选地,所述R2选自-H、1-15个碳原子的烷基或烷氧基、3-8个碳原子环烷基、2-15个碳原子的烯烃基、
本发明的一些实施方式中,优选地,所述R2选自-H、1-15个碳原子的烷基或烷氧基、3-8 个碳原子环烷基、2-15个碳原子的烯烃基、
本发明的一些实施方式中,优选地,所述R2选自1-15个碳原子的烷基或烷氧基、3-8个碳原子环烷基、2-15个碳原子的烯烃基。
在本发明的一些实施方式中,所述R3选自-H、-NO2、1-8个碳原子的烷基或烷氧基、3-6个碳原子的环烷基、2-8个碳原子的烯烃基、
在本发明的一些实施方式中,所述X‘和Y’相同或不同,各自独立地选自-S-、-S-S-、-O-、-CO-。
本发明的一些实施方式中,所述R3选自-H、1-8个碳原子的烷基或烷氧基。
本发明的一些实施方式中,所述Y1、Y2及Y3相同或不同,各自独立地选自-H、1-8个碳原子的烷基或烷氧基。
本发明的一些实施方式中,所述R3表示-H。
本发明的一些实施方式中,所述Y1、Y2及Y3表示-CH3。
本发明的一些实施方式中,通式Ⅰ的化合物选自由通式Ⅰ-1至Ⅰ-17所示化合物组成的组:
以及
其中,
所述R2选自-H、1-20个碳原子的烷基或烷氧基、3-8个碳原子的环烷基、2-20个碳原子的烯烃基或R1所示基团。
本发明还提供了一种通式Ⅰ所述的酰基肟酯类化合物的制备方法,包含如下步骤:
a、中间体Ⅰ-A的合成:以苯、二苯硫醚或硫杂蒽酮等为起始原料,与含有R2基团的酰卤化合物,在三氯化铁、三氯化铝或氯化锌等作用下,通过付克酰基化反应,合成中间体Ⅰ-A:
b、中间体Ⅰ-B的合成:中间体I在通有氯化氢或加盐酸的情况下与亚硝酸甲酯、亚硝酸乙酯或亚硝酸异戊酯等进行氧化反应,生成酰基肟中间体Ⅰ-B:
c、酰基肟酯类光引发剂合成:中间体Ⅰ-B与含有M1结构的酰卤或酸酐,在吡啶或三乙胺等缚酸剂存在下,在二氯甲烷、二氯乙烷或二氧六环等做溶剂下合成通式Ⅰ的化合物。
其中,
所述R1和R4相同或不同,各自独立地选自其中,所述R3选自-H、-NO2、1-8个碳原子的烷基或烷氧基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基、
所述Y1、Y2及Y3相同或不同,各自独立地选自-H、-CH3、2-8个碳原子的烷基或烷氧基、3-8个碳原子的环烷基、2-8个碳原子的烯烃基;
所述X‘和Y’相同或不同,各自独立地选自-S-、-S-S-、-O-、-CO-;
上述R1和R4选择的取代基中,所述环状结构中一个或多个H原子可以被F、Cl、Br、I、 OH、NO2、1-8个碳原子的烷基或烷氧基或2-8个碳原子的烯烃基取代,
所述R2选自-H、1-8个碳原子的烷基或烷氧基、3-8个碳原子的环烷基或者2-8个碳原子的烯烃基
或者与R1所示基团相同;
前提是:所述R1为时,所述R4不为
且所述R1和R4中至少一个表示
X为卤素。
具体反应路线如下:
本发明中所述的步骤a中间体I-A合成的具体操作为:氮气保护下,向有机溶剂A中加入起始原料(苯、二苯硫醚或硫杂蒽酮等)、AlCl3搅拌混合,冰盐水浴冷却至-5℃左右,滴加R2基团的酰卤化合物与有机溶剂A的混合液,温度控制在-5℃~5℃,约2h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,后处理得到白色固体中间体I-A。起始原料、AlCl3和R2基团的酰卤化合物的最佳摩尔比为1:1.1:1.05。
本发明中所述的有机溶剂A为二氯甲烷、二氯乙烷、氯仿或四氯化碳。
本发明中所述的步骤b中间体I-B合成的具体操作为:有机溶剂B中加入中间体I-A,搅拌均匀后,室温下加入盐酸或通氯化氢,通亚硝酸甲酯或滴加亚硝酸异戊酯,室温搅拌反应3~5h,减压浓缩,重结晶得白色固体中间体I-B。
本发明中所述的有机溶剂B为四氢呋喃、异丙醚、甲基叔丁基醚、乙醚、苯甲醚、丁醚、乙二醇二乙醚及二氧六环等。
本发明中所述的步骤c酰基肟酯光引发剂合成的具体操作为:向有机溶剂C中加入中间体Ⅰ-B和吡啶或三乙胺,搅拌均匀,冰盐水浴冷却至0℃左右开始滴加M1基团的酰卤化合物及 有机溶剂C的混合液,1.5h左右滴加完毕,自然恢复至室温继续搅拌反应2h左右,处理得浅黄色油状液体,即为本发明通式Ⅰ所示的酰基肟酯类化合物。
本发明中所述的有机溶剂C为二氯甲烷、二氯乙烷、氯仿、四氯化碳或二氧六环。所述的中间体Ⅰ-B与含有M1结构的酰卤或酸酐的摩尔比为1:1.1。
本发明还提供了一种通式Ⅰ所述酰基肟酯类化合物性能及在UV光固化材料中的应用。
本发明的有益效果:本发明所述的酰基肟酯类化合物在质量浓度相同的情况下,其紫外吸收谱图与OXE-1的紫外吸收谱图相同或相似,其中本发明所述的酰基肟酯类化合物的热稳定性明显比OXE-1稳定;本发明所述的酰基肟酯类化合物有部分的物质结构在紫外吸收图谱中与OXE-1有明显红移,在300~365nm有较大吸收,可实现LED冷光源作为激活光源使用,本发明所述的酰基肟酯类化合物的应用性能(感光度、热稳定性、溶解性)比现有的OXE-1的应用性能好。
同时,相比于现有同类产品,所述的酰基肟酯类化合物在整体上表现出了显著改善的综合应用性能(溶解性,稳定性、显影性、成形膜的表面抗折皱性、使用安全性),另外,本发明所述的酰基肟酯类化合物还具有非常优异的储存稳定性和很高的光固化活性,在低曝光剂量下,就能交联固化且固化效果极佳,光固化过程不产生有毒、有害物质,使用安全性高。制得的膜边缘平整无缺陷,没有浮渣,整个图案完整度好,表面没有折皱,制成的彩色滤光片光学透明度高,不漏光。突出的表现在感光性组合物的气味性、存储稳定性、显影性、成形膜的表面抗折皱性、使用安全性等方面。
附图说明
图1为(E)-2-((3-苯甲酰氧基-2,4,6-三甲基苯甲酰氧基)亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1)和OXE-1的紫外吸收线比较图,其中A为(E)-2-((3-苯甲酰氧基-2,4,6-三甲基苯甲酰氧基)亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮的紫外吸收线。
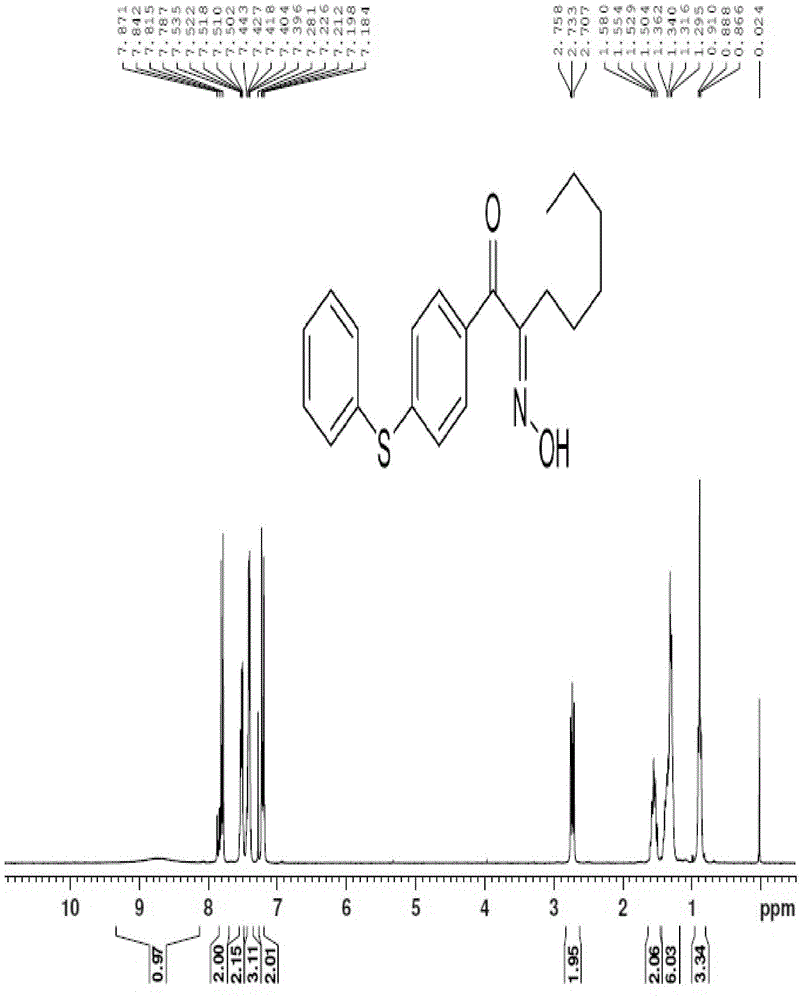
图2为(E)-2-((羟亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1-3)的核磁1HNMR谱图。
图3为(E)-2-((3-苯甲酰氧基-2,4,6-三甲基苯甲酰氧基)亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1)的核磁1HNMR谱图。
图4为(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-(羟亚氨基)辛-1-酮(化合物Ⅰ-7-1-3)的核磁1HNMR谱图。
图5为(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-((4-(苯巯基)苯甲酰氧基)亚氨基)辛-1-酮(化合物Ⅰ-7-1)的核磁1HNMR谱图。
图6为(E)-2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基)-1-(3-苯甲酰基-2,4,6-三甲基苯基)辛-1-酮(化合物Ⅰ-13-1)的核磁1HNMR谱图。
图7为1-氯-4-(2-羟亚胺基)辛酸酯基硫杂蒽酮(化合物Ⅰ-4-1-3)的核磁1HNMR谱图。
图8为(E)-1-氯-(2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基))-4-辛酸酯基硫杂蒽酮(化合物Ⅰ-4-1)的核磁1HNMR谱图。
图9为(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-((4-(苯巯基)苯甲酰氧基)亚氨基)辛-1- 酮(化合物Ⅰ-7-1)的UV吸收谱图。
图10为(E)-2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基)-1-(3-苯甲酰基-2,4,6-三甲基苯基)辛-1-酮(化合物Ⅰ-13-1)的UV吸收谱图。
图11为(E)-1-氯-(2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基))-4-辛酸酯基硫杂蒽酮(化合物Ⅰ-4-1)的UV吸收谱图。
具体实施方式
本发明通式Ⅰ的化合物优选如下化合物中的一种或多种:
实施例1
正辛酰氯(Ⅰ-2-1-1)的制备
三口烧瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加165g氯化亚砜及30ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得浅黄色溶液114g,即为化合物Ⅰ-2-1-1。
1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1-2)的制备
三口烧瓶,加入123g(0.66mol)二苯硫醚和350ml二氯甲烷,冰盐水浴降温至-5℃,通氮气,加入97.1g(0.73mol)无水A1C13,加干燥管、回流冷凝管及尾气吸收,缓慢滴113g(0.69mol)的正辛酰氯和50ml二氯甲烷的混合液,控制温度在-5~5℃,约2h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,TLC监控反应完全。将反应物缓慢倒入200ml 10%的冰的稀盐酸中,搅拌30min后分液,100ml二氯甲烷萃取水相,合并有机相,3×100ml水洗涤,2%NaHCO3溶液调至中性后分液,100ml水洗1次,无水MgSO4干燥,过滤,减压浓缩,重结晶得白色固体175g,即为化合物Ⅰ-2-1-2,产率85%。MP:31-32℃;MS:m/z=312.15。
1HNMR(300MHz,CDCl3),δ=0.87(t,J=6.8Hz,3H),1.26~1.35(m,8H),1.70(m,2H),2.89(t,J=7.4Hz,2H),7.21(d,J=8.3Hz,2H),7.38~7.42(m,3H),7.47~7.52(m,2H),7.82(d,J=8.3Hz,2H)
(E)-2-((羟亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1-3)的制备
三口烧瓶,加入300ml氯化氢的四氢呋喃溶液(含29gHCl)和31.2g(0.1mol)化合物Ⅰ-2-1-2,室温搅拌溶解,室温下滴加150ml亚硝酸甲酯的四氢呋喃溶液(0.12mol),至反应完全后,减压浓缩,得橙红色油状液体40g,加入250ml二氯甲烷,3×100ml水洗涤,无水MgSO4干燥,过滤后减压浓缩,得红棕色油状液体34g,加入120ml石油醚,搅拌下加热溶解完全,缓慢降温至-5℃,析出白色固体,抽滤,石油醚洗滤饼,得白色固体18.7g,即为化合物Ⅰ-2-1-3,产率55%。
1HNMR(300MHz,CDCl3),0.88(t,J=6.6Hz,3H),2.73(t,J=7.5Hz,2H),1.29~1.36(m,6H),1.50~1.58(m,2H),8.73(br,1H),7.18~7.28(d,J=4.2Hz,2H),7.39~7.44(m,3H),7.50~7.53(m,2H),7.78-7.87(d,J=8.7Hz,2H),如图2所示。
3-苯甲酰基-2,4,6-三甲基苯甲酰氯(化合物Ⅰ-2-1-4)的制备
三口烧瓶,加入二氯甲烷100ml、3-苯甲酰基-2,4,6-三甲基苯甲酸26.8g(0.1mol)以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加36.9g(0.3mol)氯化亚砜及50ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得红棕色油状液体30g,即为化合物Ⅰ-2-1-4。
(E)-2-((3-苯甲酰氧基-2,4,6-三甲基苯甲酰氧基)亚氨基)-1-(4-(苯巯基)苯基)辛-1-酮(化合物Ⅰ-2-1)的制备
加入6.82g(0.02mol)化合物Ⅰ-2-1-3和50ml二氯甲烷,降温至0℃左右后加入2.37g(0.03mol)吡啶,控温滴加6.29g(0.022mol)化合物Ⅰ-2-1-4及20ml二氯甲烷,滴加完毕后自然升温反应3h,反应完毕,加入30ml H20,搅拌30min后分液,加入饱和NaHCO3溶液100ml洗至pH=10左右,水洗涤至中性,再加入2%稀盐酸调至pH=4左右,3×100ml水洗至中性,无水MgSO4干燥,过滤,减压浓缩得橙红色油状液体8.3g,收率70%,即为化合物Ⅰ-2-1。
1HNMR(300MHz,CDCl3),0.81~0.89(t,J=6.0Hz,3H),2.76~2.81(t,J=8.7Hz,2H),1.22~1.32(m,4H),1.47-1.57(m,4H),2.13~2.15(m,6H),2.36~2.42(t,J=5.8Hz,3H),7.03(s,J=7.8Hz,1H),7.20~7.45(d,J=4.2Hz,2H),7.50~7.54(m,5H),7.55~7.56(m,2H), 7.73(s,J=5.7Hz,1H),7.83~7.89(d,J=8.3Hz,2H),7.98~8.01(m,2H),如图3所示。
实施例2
正辛酰氯(Ⅰ-7-1-1)的制备
三口烧瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加165g氯化亚砜及30ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得浅黄色溶液114g,即为化合物Ⅰ-7-1-1。
1-(3-苯甲酰基-2,4,6-三甲基苯基)辛-1-酮(Ⅰ-7-1-2)的制备
三口烧瓶,加入100g(0.45mol)3-苯甲酰基-2,4,6-三甲基苯和300ml二氯甲烷,冰盐水浴降温至-5℃,通氮气,加入65.3g(0.49mol)无水A1C13,加干燥管、回流冷凝管及尾气吸收,缓慢滴76.1g(0.47mol)的正辛酰氯和50ml二氯甲烷的混合液,控制温度在-5~5℃,约2h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,TLC监控反应完全。将反应物缓慢倒入200ml 10%的冰的稀盐酸中,搅拌30min后分液,100ml二氯甲烷萃取水相,合并有机相,3×100ml水洗涤,2%NaHCO3溶液调至中性后分液,100ml水洗1次,无水MgSO4干燥,过滤,减压浓缩,重结晶得棕黄色固体129g,即为化合物Ⅰ-7-1-2,产率82%。MS:m/z=350.22。
(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-(羟亚氨基)辛-1-酮(Ⅰ-7-1-3)的制备
三口烧瓶,加入300ml氯化氢的四氢呋喃溶液(含29g HCl)和35.0g(0.1mol)化合物Ⅰ-7-1-2,室温搅拌溶解,室温下滴加150ml亚硝酸甲酯的四氢呋喃溶液(0.12mol),至反应完全后,减压浓缩,得橙红色油状液体44g,加入250ml二氯甲烷,3×100ml水洗涤,无水MgSO4干燥,过滤后减压浓缩,得红棕色油状液体36g,加入120ml石油醚,搅拌下加热溶解完全,缓慢降温至-5℃,析出棕红色固体,抽滤,石油醚洗滤饼,得棕红色固体20g,即为化合物Ⅰ-7-1-3,产率53%。
1HNMR(300MHz,CDCl3),0.86~0.90(t,J=6.4Hz,3H),2.64~2.70(t,J=8.4Hz,2H),1.29~1.37(m,6H),1.49~1.54(m,2H),1.89(t,J=5.7Hz,3H),2.01~2.16(m,6H),8.99(br,1H),6.95(s,J=7.8Hz,1H),7.28~7.45(m,2H),7.56~7.61(m,1H),7.82~7.84(d,J=6.3Hz,2H),如图4所示。
2-氯-1-(4-(苯巯基)苯基)乙酮(化合物Ⅰ-7-1-4)的制备
三口烧瓶,加入50g(0.27mol)二苯硫醚和200ml二氯甲烷,冰盐水浴降温至-5℃,通氮气,加入39.4g(0.30mol)无水A1C13,加干燥管、回流冷凝管及尾气吸收,缓慢滴31.8g(0.28mol)的氯乙酰氯和50ml二氯甲烷的混合液,控制温度在-5~5℃,约1h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,TLC监控反应完全。将反应液缓慢倒入10%的冰的稀盐酸中,PH=4,搅拌30min后分液,100ml二氯甲烷萃取水相,合并有机相,3×100ml水洗涤,2%NaHCO3溶液调至中性后分液,100ml水洗1次,无水MgSO4干燥,过滤,减压浓缩,重结晶后得到45g黄色固体,即为化合物Ⅰ-7-1-4,收率:64%。MS:m/z=262.7
4-苯巯基苯甲酸(化合物Ⅰ-7-1-5)的制备
在三口瓶中,加入冷却的25%NaOH溶液,NaClO溶液(10%),三甲基苄基溴化铵,控温0℃~5℃,控温缓慢滴45g(0.17mol)的2-氯-1-(4-(苯巯基)苯基)已酮(化合物Ⅰ-7-1-4)和200ml二氯甲烷的混合液,约2h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,TLC监控反应完全。加入亚硫酸氢钠,搅拌30min后加入200ml二氯甲烷萃和10%的盐酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合并有机相,3×100ml水洗涤至中性后分液,无水MgSO4干燥,过滤,减压浓缩,重结晶后得到30g黄色固体,即为化合物Ⅰ-7-1-5,收率:76.7%。
4-(苯巯基)苯甲酰氯(化合物Ⅰ-7-1-6)的制备
三口烧瓶,加入二氯甲烷100ml、4-(苯巯基)苯甲酸21g(0.091mol)以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加11.2g(0.095mol)氯化亚砜及30ml二氯甲烷混合液,约0.5h滴加完毕,回流1h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得棕黄色油状液体26g,即为化合物Ⅰ-7-1-6。(与甲醇酯化后MS:m/z=244.3)
(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-((4-(苯巯基)苯甲酰氧基)亚氨基)辛-1-酮(化合物Ⅰ-7-1)的制备
加入7.58g(0.02mol)化合物Ⅰ-7-1-3和50ml二氯甲烷,降温至0℃左右后加入2.37g(0.03mol)吡啶,控温滴加5.45g(0.022mol)化合物Ⅰ-7-1-6及20ml二氯甲烷,滴加完毕后自然升温反应3h,反应完毕,加入30ml H20,搅拌30min后分液,加入饱和NaHCO3溶液100ml洗至pH=10左右,水洗涤至中性,再加入2%稀盐酸调至pH=4左右,3×100ml水洗至中性,无水MgSO4干燥,过滤,减压浓缩得橙黄色极粘稠液体(近固体),乙醇重结晶得8.6g,收率73%,即为化合物Ⅰ-7-1。
1HNMR(300MHz,CDCl3),0.85~0.89(t,J=6.6Hz,3H),2.82~2.87(t,J=8.7Hz,2H),1.30~1.32(m,4H),1.45(m,2H),1.60~1.65(m,2H),1.97(m,3H),2.12(m,3H),2.24(t,J=5.8Hz,3H),6.99(s,J=7.8Hz,1H),7.21~7.28(d,J=8.9Hz,2H),7.43~7.62(m,8H),7.90~7.93(m,4H),如图5所示。
实施例3
正辛酰氯(Ⅰ-13-1-1)的制备
三口烧瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加165g氯化亚砜及30ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得浅黄色溶液114g,即为化合物Ⅰ-13-1-1。
1-(3-苯甲酰基-2,4,6-三甲基苯基)辛-1-酮(Ⅰ-13-1-2)的制备
三口烧瓶,加入100g(0.45mol)3-苯甲酰基-2,4,6-三甲基苯和300ml二氯甲烷,冰盐水浴降温至-5℃,通氮气,加入65.3g(0.49mol)无水A1C13,加干燥管、回流冷凝管及尾气吸收,缓慢滴76.1g(0.47mol)的正辛酰氯和50ml二氯甲烷的混合液,控制温度在-5~5℃,约2h滴加完毕,撤去冰盐水浴,自然恢复至室温,继续搅拌反应2~3h,TLC监控反应完全。将反应物缓慢倒入200ml 10%的冰的稀盐酸中,搅拌30min后分液,100ml二氯甲烷萃取水相,合并有机相,3×100ml水洗涤,2%NaHCO3溶液调至中性后分液,100ml水洗1次,无水MgSO4干燥,过滤,减压浓缩,重结晶得棕黄色固体129g,即为化合物Ⅰ-13-1-2,产率82%。MS:m/z=350.22。
(E)-1-(3-苯甲酰基-2,4,6-三甲基苯基)-2-(羟亚氨基)辛-1-酮(Ⅰ-13-1-3)的制备
三口烧瓶,加入300ml氯化氢的四氢呋喃溶液(含29gHCl)和35.0g(0.1mol)化合物Ⅰ-13-1-2,室温搅拌溶解,室温下滴加150ml亚硝酸甲酯的四氢呋喃溶液(0.12mol),至反应完全后,减压浓缩,得橙红色油状液体44g,加入250ml二氯甲烷,3×100ml水洗涤,无水MgSO4干燥,过滤后减压浓缩,得红棕色油状液体36g,加入120ml石油醚,搅拌下加热溶解完全,缓慢降温至-5℃,析出棕红色固体,抽滤,石油醚洗滤饼,得棕红色固体20g,即为化合物Ⅰ-13-1-3,产率53%。
1HNMR(300MHz,CDCl3),0.86~0.90(t,J=6.4Hz,3H),2.64~2.70(t,J=8.4Hz,2H),1.29~1.37(m,6H),1.49~1.54(m,2H),1.89(t,J=5.7Hz,3H),2.01~2.16(m,6H),8.99(br,1H),6.95(s,J=7.8Hz,1H),7.28~7.45(m,2H),7.56~7.61(m,1H),7.82~7.84(d,J=6.3Hz,2H)。
3-苯甲酰基-2,4,6-三甲基苯甲酰氯(化合物Ⅰ-13-1-4)的制备
三口烧瓶,加入二氯甲烷100ml、3-苯甲酰基-2,4,6-三甲基苯甲酸26.8g(0.1mol)以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加36.9g(0.3mol)氯化亚砜及50ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得红棕色油状液体30g,即为化合物Ⅰ-13-1-4。
(E)-2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基)-1-(3-苯甲酰基-2,4,6-三甲基苯基)辛-1-酮(化合物Ⅰ-13-1)的制备
加入7.8g(0.02mol)化合物Ⅰ-13-1-3和50ml二氯甲烷,降温至0℃左右后加入2.37g(0.03mol)吡啶,控温滴加6.29g(0.022mol)化合物Ⅰ-13-1-4及20ml二氯甲烷,滴加完毕后自然升温反应3h,反应完毕,加入30ml H20,搅拌30min后分液,加入饱和NaHCO3溶液100ml洗至pH=10左右,水洗涤至中性,再加入2%稀盐酸调至pH=4左右,3×100ml水洗至中性,无水MgSO4干燥,过滤,减压浓缩得浅黄色略带粘稠固体,乙醇重结晶得9.8g,收率78%,即为化合物Ⅰ-13-1。
1HNMR(300MHz,CDCl3),0.83~0.87(t,J=6.6Hz,3H),2.72~2.77(t,J=8.7Hz,2H),1.26~1.37(m,4H),1.50-1.63(m,4H),1.92~2.05(m,3H),2.12~2.16(t,J=12.4Hz,3H),2.30(m, 6H),2.72~2.77(m,6H),6.95-6.97(d,J=8.1Hz,2H),7.45~7.57(m,4H),7.60-7.64(m,2H),7.80~7.88(m,4H),如图6所示。
实施例4
正辛酰氯(化合物Ⅰ-4-1-1)的制备
三口烧瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加165g氯化亚砜及30ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得浅黄色溶液114g,即为化合物Ⅰ-4-1-1。
1-氯-4-辛酸酯基硫杂蒽酮(化合物Ⅰ-4-1-2)的制备
加入5.24g(0.02mol)化合物1-氯-4-羟基硫杂蒽酮和50ml二氯甲烷,降温至0℃左右后加入2.37g(0.03mol)吡啶,控温滴加3.56g(0.022mol)化合物Ⅰ-4-1-1及20ml二氯甲烷,滴加完毕后自然升温反应3h,TLC板跟踪至反应完毕,加入30ml H20,搅拌30min后分液,加入饱和NaHCO3溶液100ml洗至pH=10左右,水洗涤至中性,再加入2%稀盐酸调至pH=4左右,3×100ml水洗至中性,无水MgSO4干燥,过滤,减压浓缩得棕黄色固体,乙醇重结晶得6.5g,收率84%,即为化合物Ⅰ-4-1-2。
1-氯-4-(2-羟亚胺基)辛酸酯基硫杂蒽酮(Ⅰ-4-1-3)的制备
三口烧瓶,加入300ml氯化氢的四氢呋喃溶液(含29gHCl)和38.9g(0.1mol)化合物Ⅰ-4-1-2,室温搅拌溶解,室温下滴加150ml亚硝酸甲酯的四氢呋喃溶液(0.12mol),至反应完全后,减压浓缩,得橙红色油状液体49g,加入250ml二氯甲烷,3×100ml水洗涤,无水MgSO4干燥,过滤后减压浓缩,得红棕色油状液体41g,加入120ml石油醚,搅拌下加热溶解完全,缓慢降温至-5℃,析出棕土黄色固体,抽滤,石油醚洗滤饼,得土黄色固体26.7g,即为化合物Ⅰ-4-1-3,产率64%。
1HNMR(300MHz,CDCl3),0.85~0.88(t,J=7.3Hz,3H),2.71~2.75(t,J=9.4Hz,2H),1.30~1.36(m,6H),1.50~1.57(m,2H),11.24(br,1H),7.21(s,J=7.2Hz,1H),7.26~7.31(s, J=7.2Hz,1H),7.46(s,J=8.1Hz,1H),7.54~7.57(d,J=8.9Hz,2H),8.24-8.26(s,J=7.2Hz,1H),如图7所示。
3-苯甲酰基-2,4,6-三甲基苯甲酰氯(化合物Ⅰ-4-1-4)的制备
三口烧瓶,加入二氯甲烷100ml、3-苯甲酰基-2,4,6-三甲基苯甲酸26.8g(0.1mol)以及2滴DMF,搭上回流冷凝管、恒压滴液漏斗、干燥管及碱液尾气吸收装置,加热至回流,缓慢滴加36.9g(0.3mol)氯化亚砜及50ml二氯甲烷混合液,约1h滴加完毕,回流2h,减压浓缩,加入新鲜二氯甲烷100ml再次减压浓缩,得红棕色油状液体30g,即为化合物Ⅰ-4-1-4。
(E)-1-氯-(2-(3-苯甲酰基-2,4,6-三甲基苯甲酰亚氨酯基))-4-辛酸酯基硫杂蒽酮(化合物Ⅰ-13-1)的制备
加入8.36g(0.02mol)化合物Ⅰ-4-1-3和50ml二氯甲烷,降温至0℃左右后加入2.37g(0.03mol)吡啶,控温滴加6.29g(0.022mol)化合物Ⅰ-4-1-4及20ml二氯甲烷,滴加完毕后自然升温反应3h,反应完毕,加入30ml H20,搅拌30min后分液,加入饱和NaHCO3溶液100ml洗至pH=10左右,水洗涤至中性,再加入2%稀盐酸调至pH=4左右,3×100ml水洗至中性,无水MgSO4干燥,过滤,减压浓缩得土黄色固体,乙醇重结晶得11.3g,收率85%,即为化合物Ⅰ-4-1。MP:140.76℃。
1HNMR(300MHz,CDCl3),0.81~0.83(t,J=6.5Hz,3H),2.76~2.78(t,J=8.5Hz,2H),1.21~1.26(m,4H),1.47~1.52(m,4H),2.13~2.15(m,6H),2.42(m,3H),7.23(s,J=7.4Hz,1H),7.48~7.56(m,8H),7.85~7.88(m,2H),8.73~8.46(s,J=8.4Hz,1H),如图8所示。
应用实施例1
检测了化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1在常用的有机溶剂,如乙酸乙酯、乙二醇单乙醚、丙二醇甲醚、丙二醇甲醚醋酸酯中的溶解性。分别取四份等量的化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1置于四个试样瓶中,分别用乙二醇单乙醚、丙二醇甲醚醋酸酯溶解,通过TLC检测光引发剂在溶液中的稳定性。结果显示均具有一定的溶解性,且此类光引发剂在有机溶剂中的稳定性比较好,避光条件下在常用溶剂中均能保持20天以上不发生分解。
应用实施例2
利用差热-热重分析仪测定化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1的热解性能。升温速度: 10℃/min。检测了化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1初始分解温度均在150℃以上,热稳定性较好。
应用实施例3
将化合物化合物Ⅰ-2-1,Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1溶解在乙腈中,分别配成一定摩尔浓度的溶液,通过用紫外-可见分光光度计测其紫外吸收谱图,进行比较。通过检测谱图可以看出所合成的几种化合物紫外吸收谱带较宽泛,在300~365nm均有较大吸收,如图1,图9、图10、图11所示。
应用实施例4
利用实时红外测试手段,比较了实施例中化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1引发甲基丙烯酸羟乙酯聚合的双键转化率。以丙酮为溶剂,配制化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1的浓度为单体浓度的3%的试样,涂于KBr盐片上,然后放入Nico-let5700中,用紫外光点光源照射样品,调节样品表面的紫外光光强为30mW/cm2。单体的双键转化率用近红外实时采集,实时红外参数设置为数据采集间隔0.3985s,每个光谱扫描1次,分辨率为4cm-1。甲基丙烯酸羟乙酯在近红外谱图中碳碳双键的特征吸收峰在1630cm-1处,随着光固化反应的进行,碳碳双键变成碳碳单键,双键的吸收峰强度随光照时间增加而变弱,所以利用碳碳双键的特征吸收峰的变化来反映其聚合反应的变化程度。双键转化率(DC)由数据处理软件结合下式计算得到。
DC(%)=[1-(At/Ao)]×100%
式中Ao和At分别为样品在固化前和光照后t时刻在1630cm-1处甲基丙烯酸羟乙酯双键特征吸收峰的面积。检测结果显示化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1均可以引发甲基丙烯酸羟乙酯聚合,在光照l0min之后,均能使丙烯酸双键的转化率达60%以上。
应用实施例5
用聚乙烯吡咯烷酮(MW=40000)与聚甲基丙烯酸酯树脂(Mn=50000)作成膜树脂,2-羟丁基丙烯酸酯作聚合单体,分别加入化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1,加入适量的链转移剂和染料,用丙二醇甲醚作溶剂,配成自由基聚合成像感光胶液1、2、3、4。将以上胶液按以下方法进行性能测试。将感光胶液1~4用离心机旋涂在预先处理好的并满足下列条件的PS铝版基上,铝板基尺寸:1030mm×800mm;铝板基厚度:0.28~0.3mm;砂目规格:Ra=0.5~0.6μm,Rh=0.3~0.35μm;阳极氧化膜重量:3~3.5g/m2。控制离心涂布机的转速,使涂在铝版基上的涂布量(以固含量计)为0.5~2.5g/m2,在离心涂布机上初步干燥后,转移到100℃的鼓风干燥机中干燥3min,得紫激光CTP原版。将原版经紫激光曝光,用Ugra测试条做掩膜测试版材的感光性能。曝光后,用1%NaOH水溶液显影。曝光区,可光聚合化合物在引发剂存在下发生聚合反应,显影液中不溶,而非曝光区是可溶的,于是得到阴图。通过曝光显影,从得到的图像的的连续调梯尺评价感度,从微线条测试块区域评价精度,从而评价感光组合物感光性能的优劣。实验结果显示在曝光时间均为40s的情况下,化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1的版材均能显到连续调梯尺的3段以上,精度在6μ以上。由此可见,化合物Ⅰ-2-1、Ⅰ-7-1、Ⅰ-13-1及Ⅰ-4-1在一定曝光时间及曝光量下,能达到较好的成像效果,可适用于紫激光成像体系中。
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用上述揭示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!