一种RNA反义纯化方法与流程
本发明涉及生物技术领域,尤其涉及一种RNA反义纯化方法。
背景技术:
长链非编码RNA(Long non-coding RNA,lncRNA)是长达两百个核苷酸以上的转录本,绝大多数位于细胞核内。虽然lncRNA没有编码任何蛋白质,但它们在不同组织和发育阶段的表达依然具有特异性,这说明lncRNA具有重要的生物学意义。在测序技术的帮助下,人们已经发现了数千种lncRNA,但这些lncRNA的生物学功能依然还是个谜。
2013年,Jesse M.Engreitz教授开发了RNA反义纯化(RNAantisensePurification,RAP)技术,该技术可以在转录后水平鉴定RNA与RNA及相关DNA和蛋白质的互作。其原理如下:首先用紫外交联固定细胞,以维持如lncRNA等调控RNA与RNA、RBP的相互作用,然后进行细胞裂解,接着用生物素标记的寡核苷酸探针组与靶lncRNA杂交,基于生物素和链霉亲和素相互作用的原理,用链霉亲和素磁珠来分离、纯化探针-基因或蛋白质复合体,最后从纯化的复合体中分离蛋白质或RNA,以进行下游的分析。
RAP技术被不同领域的研究者们广泛采用。如Engreitz J M等(2013)利用RAP技术通过研究LncRNA Xist成功地阐释了Xist使X染色体失活的作用机理(Engreitz J M,Pandyajones A,Mcdonel P,et al.The Xist lncRNA exploits three-dimensional genome architecture to spread across the X-chromosome[J].Science,2013,341(6147):L35-L38.)。随着人们进一步认识非编码RNA在转录后水平发挥的重要作用,研究者对RAP技术进行不断的完善,旨在阐明lncRNA在机体内与内源性RNA、蛋白质的互作关系。2014年,Engreitz J M及其同事们运用RAP技术研究了核内小RNA U1和lncRNA Malat1与前体mRNA互作的作用机理。通过使用不同交联试剂及交联条件,改进的RAP技术能够进一步研究体内RNA-RNA互作关系及RNA在染色体上的定位及参与的转录调控(Engreitz J,Sirokman K,Mcdonel P,et al.RNA-RNA Interactions Enable Specific Targeting of Noncoding RNAs to Nascent Pre-mRNAs and Chromatin Sites[J].Cell,2014,159(1):188-99.)。
然而,现有RAP技术因磁珠会吸附非特异性的核酸及蛋白质、探针组与RNA杂交的效率不高、核酸及蛋白质等产品回收效率不高等问题而存在特异性低、灵敏度不佳的缺点。
技术实现要素:
有鉴于此,有必要针对上述的问题,提供一种RNA反义纯化方法。
为了实现上述目的,本发明采用如下技术方案:
一种RNA反义纯化方法,包含以下步骤:
S1、细胞的交联、裂解及基因组DNA的消化,得到RNA样品;
S2、探针组溶液的制备:将生物素标记的各探针溶解后混合,于85℃变性3min,快速转移冰浴,得到探针组溶液;
S3、磁珠预处理:使用BSA(牛血清白蛋白)和寡核苷酸随机引物封闭链霉亲和素标记的磁珠;
S4、探针组-磁珠复合物的制备:将探针组溶液加入到预处理后的磁珠中,得到探针组-磁珠复合物;
S5、反义纯化:向RNA样品中加入探针组-磁珠复合物,65℃变性10min,37℃、42℃和45℃各洗涤2次,每次洗涤5min,得到RNA-探针组-磁珠复合物;
S6、RNA的洗脱:将RNA-探针组-磁珠复合物加入RNA ElutionBuffer混匀,95℃加热2min,收集上清,向上清中加入Proteinase K Buffer和Proteinase K,温育消化得到洗脱后的RNA;
S7、RNA的纯化:向洗脱后的RNA中加入等洗脱体积的苯酚-氯仿-异戊醇混合液混匀,离心收集上层水相,向水相中加入5M NaCl溶液、glycogen和无水乙醇,混匀后-80℃沉淀1~16h,离心弃上清,加入预冷的80%乙醇,离心弃上清,室温静置风干,加入无RNase水,65℃温育15min。
优选地,步骤S1中取约1×108个细胞,去除培养基后用PBS洗涤一次,进行紫外交联,向交联后细胞中加入15ml预冷的PBS并短暂置于冰上,收集细胞,4℃400g离心3min收集细胞沉淀;向细胞沉淀中加入1.4ml预冷的Lysis Buffer、5μl RNase Inhibitor,充分吹打裂解,抽动混匀3次,冰浴静置裂解10min,向裂解液中加入4.8μl DNase Salt Stock、15μl DNase,37℃温育10min;转移样品到冰浴环境,快速加入20μl 0.5M EDTA、10μl 0.5M EGTA、5μl 1M DTT并充分混匀;加入等体积2×Hybridization Buffer,充分混匀后冰上静置10min;4℃16000g离心10min,转移上清至新无RNase离心管中,-80℃保存备用。
优选地,步骤S2中每条探针用1×TES配制成100倍nmol的探针溶液,将所有探针溶液混合,取5μl混合液变性处理。
优选地,步骤S3磁珠预处理的方法为:用1ml 10mM Tris-HCl pH7.5重悬磁珠,颠倒混匀后磁力架上收集磁珠去上清,再重复洗涤2次;用1ml1×Hybridization Buffer重悬磁珠,颠倒混匀后磁力架上收集磁珠去上清;再向磁珠中加入1ml含BSA和寡核苷酸随机引物的1×TES,室温孵育30min后去除TES溶液。
优选地,步骤S3中TES中BSA的浓度为0.05~0.5wt%,寡核苷酸随机引物的浓度为10~20μmol/L。
优选地,步骤S3中寡核苷酸随机引物为20bp以内的DNA序列。
优选地,步骤S4制备探针-磁珠复合物的方法为:将探针组溶液加入预处理后的磁珠中,并加入100μl 2×TES,25℃温和旋转混合30min,磁力架上收集磁珠去上清;用0.35ml 1×TES洗涤磁珠两次,去除TES。
优选地,步骤S5中向RNA样品中加入探针组-磁珠复合物,65℃温育变性10min,用含0.5mM PMSF的Wash Buffer 37℃、42℃、45℃各洗涤2次,每次洗涤5min并磁力架上静置1min收集磁珠弃上清;用500μl Wash Buffer悬浮磁珠,35℃摇动洗涤2~4min,磁力架上收集磁珠并去上清,并重复洗涤3~6次,得到RNA-探针组-磁珠复合物。
优选地,步骤S6中向RNA-探针组-磁珠复合物中加入50μl RNA Elution Buffer充分混匀后,95℃加热2min,磁力架上收集磁珠并转移上清至新无RNase离心管中,重复上述步骤一次;再向上清中加入25μl 5×Proteinase K buffer、1μg Proteinase K,50℃温育消化1h。
优选地,步骤S7中RNA的纯化方法为:向洗脱后的RNA样品中加入等洗脱体积的苯酚-氯仿-异戊醇混合液,颠倒混匀15s;13000g 4℃离心10min,收集上层水相,转移到新无RNA酶离心管中;向水相中加入20μl 5M NaCl、2μl Glycogen、600μl无水乙醇,充分颠倒混匀后-80℃沉淀1~16h;4℃16100g离心30min,弃上清;加入1ml预冷的80%乙醇,4℃16100g离心10min,弃上清;室温静置风干10min;加入20~30μl RNase-free water,65℃温育15min。
与现有技术相比,本发明具有如下有益效果:
①本发明通过预先使用BSA和寡核苷酸随机引物封闭磁珠,降低了磁珠的非特异性吸附。BSA和寡核苷酸随机引物的大小正合适,既能封闭磁珠增强实验的特异性,又不会过大而影响探针与RNA的结合。
②本发明中磁珠首先与探针孵育,去除多余探针后再与RNA孵育,过量探针封闭链霉亲和素与蛋白质、核酸等的非特异性结合,提高检测特异性,灵敏度高;杂交后经梯度温度多次洗涤,保证磁珠充分结合探针并结合目标RNA,提高杂交效率。
附图说明
图1为实施例1中纯化RNA样品的琼脂糖凝胶电泳图。泳道1为Maker,泳道2为纯化后的RNA样品。
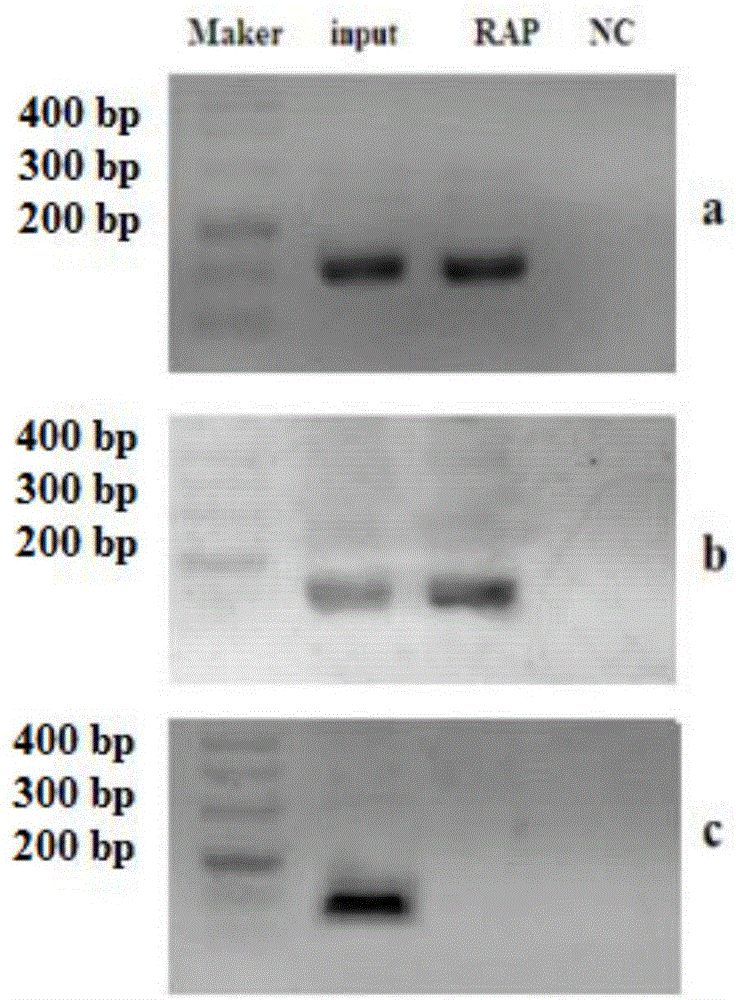
图2为实施例1中检测miRNA的琼脂糖凝胶电泳图。图a为本发明结果图,图b为对比例1实验结果图,图c为对比例2实验结果图。泳道1为Maker,泳道2为input组(10%);泳道3为实验组;泳道4为NC组。
图3为实施例2中纯化RNA样品的琼脂糖凝胶电泳图。泳道1为Maker,泳道2为纯化后的RNA样品。
图4为实施例2中检测lincRNA的琼脂糖凝胶电泳图。图a为本发明结果图,图b为对比例1实验结果图,图c为对比例2实验结果图。泳道1为Maker,泳道2为input组(10%);泳道3为实验组;泳道4为NC组。
具体实施方式
为了更好的说明本发明,下面结合附图和具体实施方式做进一步说明。本发明中所用试剂或仪器均可由市场购得,使用的检测方法等都是本领域所熟知的,在此不再赘述。
实施例1
CTNNB1为编码catenin beta 1蛋白的mRNA,物种为人类。针对CTNNB1 mRNA开展RAP实验,靶基因为hsa-miR-214。探针的设计和用生物素标记方法是本领域技术人员所熟知的,在此不再赘述。链霉亲和素偶联在磁珠(BeaverBeadsTM Streptavidin,海狸生物科技有限公司)上,并和脱硫生物素亲和结合;RNA与磁珠-RNA探针孵育,经过洗涤可以将非特异性结合RNA等去除;最后,再用洗脱液(针对链霉亲和素)进行洗脱,纯化后可以验证RNA类型。具体步骤如下:
S1、细胞的交联、裂解及基因组DNA的消化,得到RNA样品
S11、培养和收集细胞
本实施例中使用cCL-RAP细胞培养或PAR-CL细胞培养方式。采用15cm细胞培养皿培养,cCL-RAP细胞的培养使用含10v/v%FCS DMEM,PAR-CL细胞的培养使用含100μM 4SU及10v/v%FCS DMEM。培养结束时每皿约2×107个细胞,每次实验需要5皿细胞,共计约1×108个细胞。
S12、交联细胞
去除培养基后,向培养皿中加入0.3ml PBS,摇动洗涤一次后,充分去除残余PBS并将培养皿置于冰上;将培养皿置于紫外灯10~15cm距离,0.15J cm2254nm UV交联约1min(cCL),或者365nm UV交联约1min(4SU-labeled,PAR-CL)。交联结束后,尽快向培养皿中加入15ml预冷的PBS并短暂静置于冰上;用细胞刮收集细胞;4℃400g离心3min收集细胞沉淀,去除上清。
S13、裂解细胞
向细胞沉淀中加入1.4ml预冷的Lysis buffer、5μl RNase Inhibitor,并充分吹打充分裂解;使用0.4mm针头口径注射器,抽动混匀3次,冰浴静置裂解10min;裂解样品立即使用或-80℃保存一周内使用。
S14、消化基因组DNA
向细胞裂解液中添加4.8μl 1×DNase salt stock、15μl DNase(20U),37℃温育10min;转移样品到冰浴环境,快速加入20μl 0.5M EDTA、10μl 0.5M EGTA、5μl 1M DTT并充分混匀;将细胞裂解液按0.5ml、0.5ml、0.2ml及0.05ml分为四管,分别为标示实验组1样品、实验组2样品(平行试验)、NC组样品和input组样品;向NC组和各实验组样品中分别加入等体积2×Hybridization Buffer,充分混匀后冰上静置10min;4℃16,000g离心10min转移上清至新无RNase离心管中;NC组、input组和各实验组样品-80℃保存备用。
S2、探针组溶液的制备:将生物素标记的各探针溶解后混合,于85℃变性3min,快速转移冰浴,得到探针组溶液。本实施例中,实验组的探针组含有8条探针,NC组的探针组含有3条探针。具体序列如表1和表2所示。
表1、实验组用探针序列
表2、NC组用探针序列
以实验组为例说明探针组溶液的制备:向各条探针中加入100倍nmol数μl的1×TE,假如探针为1nmol的粉末,则加入100μl的TE,充分涡旋混匀后微离心后,都转移到同一离心管中,加入1×TE调整每条探针浓度为10pmol/μl,得到探针混合液;取5μl探针混合液,其中每条探针含量为50pmol,探针总量为n×50pmol,n为探针数目;85℃变性3min后,快速转移冰浴,得到实验组用探针组溶液。采用相同的方法可以制备NC组用探针组溶液。
S3、磁珠预处理:使用BSA和寡核苷酸随机引物封闭链霉亲和素标记的磁珠。
磁珠浓度为400pmol/100μl,每1pmol探针需要1pmol磁珠,按n(探针数目)×100μl/(400pmol/50pmol)链霉亲和素磁珠标准,为实验组准备磁珠,磁力架上收集磁珠并去上清;加入1ml 10mM Tris-HCl pH 7.5充分重悬后,颠倒混匀10次,磁力架上收集磁珠并去上清,重复洗涤2次;加入1ml 1×Hybridization Buffer充分重悬后,颠倒混匀10次,磁力架收集磁珠并去上清。向磁珠加入1ml含0.05~0.5wt%BSA和10~20μmol/L寡核苷酸随机引物的1×TES,室温孵育30min后置于磁力架上去除TES溶液。采用相同的方法可以制备NC组用磁珠。
S4、探针组-磁珠复合物的制备:将探针组溶液加入到预处理后的磁珠中,得到探针组-磁珠复合物。
将制备的实验组探针组溶液加入实验组用预处理后的磁珠中,并加入100μl 2×TES,25℃旋转温和混合30min,磁力架上收集磁珠并去上清,加入0.35ml 1×TES,温和旋转洗涤磁珠,置于磁力架上去除洗涤液,重复洗涤一次,得到实验组用探针组-磁珠复合物。采用相同的方法制备NC组用探针组-磁珠复合物。
S5、反义纯化
实验组样品中加入准备的实验组用探针组-磁珠复合物;垂直混匀器上65℃温育变性10min,37℃孵育杂交180min,37℃洗涤2次、42℃洗涤2次、45℃洗涤2次,每次垂直混匀器摇动洗涤5min,洗涤用Wash Buffer,在使用前预热到相应洗涤温度并加入0.5mM PMSF,洗涤后在磁力架上静置1min收集磁珠弃上清;最后一次洗涤后加入500μl Wash Buffer悬浮磁珠,35℃摇动洗涤2~4min后磁力架上收集磁珠并去上清,并重复3~6次,得到实验组用RNA-探针组-磁珠复合物。采用相同的方法制备NC组用RNA-探针组-磁珠复合物。
S6、RNA的洗脱
实验组用RNA-探针组-磁珠复合物样品磁珠加入50μl RNA Elution Buffer充分混匀后,95℃加热2min;磁力架收集磁珠并转移上清至新无RNase离心管中,重复上述步骤一次;加入25μl 5×Proteinase K buffer、1μg Proteinase K,50℃温育消化1h,得到实验组用洗脱后的RNA样品。采用相同的方法洗脱NC组用RNA-探针组-磁珠复合物,得到NC组用洗脱后的RNA样品。
向input组样品中加入25μl 5×Proteinase K buffer、1μg Proteinase K、50μl RNA Elution Buffer,50℃温育消化1h,得到input组用RNA样品。
S7、RNA的纯化
向实验组用洗脱后的RNA样品中加入等洗脱体积的(125μl)苯酚-氯仿-异戊醇混合液,颠倒混匀15s;13000g 4℃离心10min,收集上层水相并转移到新无RNA酶离心管中;加入20μl 5M NaCl、2μl Glycogen、600μl无水乙醇,充分颠倒混匀后-80℃沉淀1~16h;4℃16100g离心30min,弃上清;加入1ml预冷的80%乙醇,4℃16100g离心10min,弃上清;室温静置风干10min;加入20~30μl RNase-free water,冰上静置保存;65℃温育15min,充分溶解RNA及去除残余DNA酶活性,得到纯化后的实验组样品。采用相同的方法处理NC组用洗脱后的RNA样品和input组用RNA样品,分别得到纯化后的NC组样品和input组样品。
对纯化后的RNA样品进行了miRNA检测,具体方法如下。取5μl RNA样品开展1.5%琼脂糖凝胶电泳检测RNA富集效果,结果显示检测到明显的RNA富集(如图1)。取2μl RNA样品Agilent 2100检测RNA浓度及片段大小范围,检测结果显示富集得到的RNA浓度为6.9ng/μl,大小范围为200~700bp,均在正常范围之内。取2μl RNA样品nanodrop检测RNA纯度及浓度,结果显示其纯度为:OD260/280为1.91,浓度为8.9ng/μl,两次检测结果相近,说明已成功完成RNA的富集及纯化。
为了更好的说明本发明的效果,发明人还进行了两组对照,对照组1采用传统方法(Jesse Engreitz.RNA Antisense Purification(RAP):Experimental Protocols.2014.)检测has-MiR-214基因;对照组2采用本发明RAP试剂盒检测内参U6基因。
使用QiagenmiScript Reverse Transcription Kit(218061)逆转录RNA样品,具体方法参考逆转录试剂盒说明书;参考2×Taq PCR试剂盒说明书配置20μl PCR扩增体系,进行PCR扩增,取5~10μl PCR扩增产物开展琼脂糖凝胶电泳(2%~3%),结果如图2所示。靶基因hsa-miR-214扩增片段长度为约90bp(图2a),与对照组1(图2b)的结果相比,均处于正确范围,但比传统方法有更好的浓度和纯度。F引物序列为ACAGCAGGCACAGACAGGCAG(SEQ ID NO:12),R引物为试剂盒自带引物。对照组2(图2c)中U6扩增片段长度为94bp,F引物序列为:CTCGCTTCGGCAGCACA(SEQ ID NO:13);R引物序列为:AACGCTTCACGAATTTGCGT(SEQ ID NO:14)。
实施例2
SENP1为编码SUMO1/sentrin specific peptidase 1蛋白的mRNA,物种为人类。针对SENP1mRNA开展RAP实验,靶基因为lincRNA-p21。
S1、细胞的交联、裂解及基因组DNA的消化,得到RNA样品。具体方法与实施例1相同。
S2、探针组溶液的制备:除实验组用探针组的序列不同外,本实施例中探针组溶液制备的具体方法与实施例1相同。
本实施例中,实验组的探针组含有10条探针,NC组的探针组含有3条探针(与实施例1相同)。具体序列如表3所示。
表1、实验组用探针序列
S3、磁珠预处理:与实施例1相同。
S4、探针组-磁珠复合物的制备:与实施例1相同。
S5、反义纯化:与实施例1相同。
S6、RNA的洗脱:与实施例1相同。S7、RNA的纯化:与实施例1相同。
为了更好的说明本发明的效果,发明人还进行了两组对照,对照组1采用传统方法(Jesse Engreitz.RNA Antisense Purification(RAP):Experimental Protocols.2014.)检测lincRNA-p21基因;对照组2采用本发明RAP试剂盒检测GAPDH基因。
对纯化后的RNA样品进行了lincRNA检测,具体方法如下。取5μl RNA样品开展1.5%琼脂糖凝胶电泳检测RNA富集效果,结果显示检测到明显的RNA富集(结果如图3所示);取2μl RNA样品Agilent 2100检测RNA浓度及片段大小范围,检测结果显示富集得到的RNA浓度为8.9ng/μl,大小范围为500~3000bp均在正常范围之内;取2μl RNA样品nanodrop检测RNA纯度及浓度,结果显示其纯度为:OD260/280为1.94,浓度为10.2ng/μl,两次检测结果均证明已成功完成RNA的富集及纯化。
使用(TaKaRa PrimeScript II 1st Strand cDNA Synthesis Kit(D6210A))逆转录RNA样品,具体方法参考逆转录试剂盒说明书;参考2×Taq PCR试剂盒说明书配置20μl PCR扩增体系,进行PCR扩增,取5~10μl扩增产物开展琼脂糖凝胶电泳(2%~3%),结果如图4所示。靶基因lincRNA-p21扩增片段长度为156bp(图4a),与传统方法(图4b)的相比,结果均处于正确范围,但比传统方法有更加清晰的条带,浓度和纯度更佳,引物序列为:F为TGCAGAGCGTTTTGTTTGTC(SEQ ID NO:25);R为CAGCCTCTGGGAAGAAAATG(SEQ ID NO:26)。对照GAPDH扩增片段长度为189bp(图4c),引物序列为:F为CGGCTACTAGCGGTTTTACG(SEQ ID NO:27);R为AAGAAGATGCGGCTGACTGT(SEQ ID NO:28)。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
序列表
<110>广州伯信生物科技有限公司
<120>一种RNA反义纯化方法
<160>28
<170>PatentIn version 3.3
<210>1
<211>71
<213>人工合成
<400>1
ccatcaaatc agcttgagta gccattgtcc acgctggatt ttcaaaacag ttgtatggta 60
tacttcaaat a 71
<210>2
<211>68
<213>人工合成
<400>2
tttcagcatc tgtgatggtt cagccaaacg ctggacatta gtgggatgag cagcatcaaa 60
ctgtgtag 68
<210>3
<211>67
<213>人工合成
<400>3
gccataagct aaaatttgaa ggcagtctgt cgtaatagcc aagaatttaa catttgtttt 60
gttgagc 67
<210>4
<211>69
<213>人工合成
<400>4
gaagctgaac aagagtccca aggagacctt ccatcccttc ctgtttagtt gcagcatctg 60
aaagattcc 69
<210>5
<211>67
<213>人工合成
<400>5
gttttcaatg ggagaataaa gcagctgcac aaacaatgga atggtattta gtcctctgat 60
aacaatt 67
<210>6
<211>70
<213>人工合成
<400>6
gatatccaag gggttctccc tgggcaccaa tatcaagtcc aagatcagca gtctcattcc 60
aagccattgg 70
<210>7
<211>69
<213>人工合成
<400>7
gatccattct tgtgcattct tcacttcttg agtcactccc aaaatccatt tgtattgtta 60
ctcctcgac 69
<210>8
<211>67
<213>人工合成
<400>8
aaactgtcca aaacaaggtt ccaaataaca cctcttactg atttacccta cccatacata 60
tcccaaa 67
<210>9
<211>68
<213>人工合成
<400>9
caaacggcgg attgaccgta atgggatagg tcacgttggt gtagatgggc gcatcgtaac 60
cgtgcatc 68
<210>10
<211>67
<213>人工合成
<400>10
caccacatac aggccgtagc ggtcgcacag cgtgtaccac agcggatggt tcggataatg 60
cgaacag 67
<210>11
<211>67
<213>人工合成
<400>11
ccaatccgcg ccggatgcgg tgtatcgctc gccacttcaa catcaacggt aatcgccatt 60
tgaccac 67
<210>12
<211>21
<213>人工合成
<400>12
acagcaggca cagacaggca g 21
<210>13
<211>17
<213>人工合成
<400>13
ctcgcttcgg cagcaca 17
<210>14
<211>20
<213>人工合成
<400>14
aacgcttcac gaatttgcgt 20
<210>15
<211>67
<213>人工合成
<400>15
tttctgatgg tggcactagc gatatcacat ctcagggaga taccacagag taggagagtt 60
ggagaaa 67
<210>16
<211>70
<213>人工合成
<400>16
gtgagtcttt caaagtattt ttactggaac taagacatcg agacaggtgt aaaggaaaat 60
gtgtggtggg 70
<210>17
<211>68
<213>人工合成
<400>17
aagtagaact gggagttgga gtctgggaat ctttcacttt cagtaaaatc acagagtctg 60
atccttca 68
<210>18
<211>67
<213>人工合成
<400>18
aagcccttct ctttacttcg ctccatcagc atattcatgt agaaattgat gatctcatca 60
ttgagcc 67
<210>19
<211>71
<213>人工合成
<400>19
agagtattct gcaggcttca ttgtttatcc cacccatgga gtcgtaatag gtaatattct 60
tctttctaaa g 71
<210>20
<211>67
<213>人工合成
<400>20
tagacaacaa agagctggtc ccccacatgg tcaaggtctg ctaagtgaga cagtcttcac 60
aagagtt 67
<210>21
<211>68
<213>人工合成
<400>21
ctggtggttt ttatccctac cttttgccca tgccctttcc tacttcttag taaggcctaa 60
caacaaga 68
<210>22
<211>69
<213>人工合成
<400>22
aaacacccta ctcatagggg ctgggatctc tgtgtgctgg aatttggaaa atggaaataa 60
atctacagg 69
<210>23
<211>71
<213>人工合成
<400>23
actaaacgag cttgtataaa acagtaaaat tttcaaataa gcccagggaa aaagcacaat 60
gctggaaaga a 71
<210>24
<211>68
<213>人工合成
<400>24
cacctttcaa gttgttgctt acatacagta aataccaaga accagacaat attccaggag 60
tcctgcaa 68
<210>25
<211>20
<213>人工合成
<400>25
tgcagagcgt tttgtttgtc 20
<210>26
<211>20
<213>人工合成
<400>26
cagcctctgg gaagaaaatg 20
<210>27
<211>20
<213>人工合成
<400>27
cggctactag cggttttacg 20
<210>28
<211>20
<213>人工合成
<400>28
aagaagatgc ggctgactgt 20
- 还没有人留言评论。精彩留言会获得点赞!