ctDNA超低频突变检测文库的构建方法、试剂盒及文库检测数据的分析方法与流程
本发明涉及生物
技术领域:
,具体而言,涉及一种ctDNA超低频突变检测文库的构建方法、试剂盒及文库检测数据的分析方法。
背景技术:
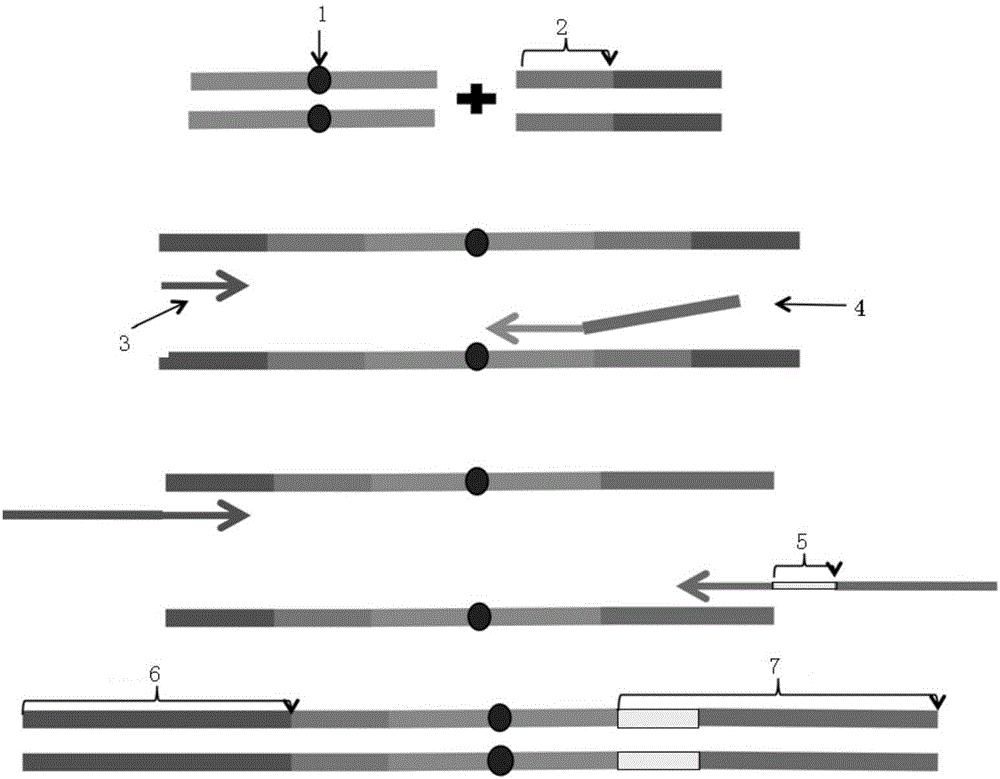
:ctDNA(circulatingtumorDNA),即循环肿瘤DNA,是由肿瘤细胞释放到血液循环系统中的DNA,它们携带有肿瘤细胞中所有的基因突变信息,因此,理论上可以通过检测ctDNA的突变状态来反应肿瘤组织的突变状态。因为无论是原发部位肿瘤还是转移部位肿瘤,均在持续不断的向血液中释放肿瘤DNA,所以ctDNA体现的是患者整体的肿瘤基因突变情况,因而更加全面;仅仅需要抽取一管血液就可以进行检测的便利性,也使之克服了肿瘤组织难以取样和不方便长期监测的问题。因此,基于血液的无创或者微创的肿瘤基因检测技术备受关注,有着广阔的应用前景。但是,在能够获得大规模临床应用之前,ctDNA检测技术还有几个技术瓶颈需要克服:1)人体正常细胞的凋亡,也会释放大量的游离DNA到血液中,这些游离DNA为ctDNA的检测制造了大量的背景噪音。一般认为,血浆中ctDNA占游离DNA的比例从0.1%到93%不等,因此,ctDNA检测技术需要克服的第一个瓶颈是超低频突变的检出。2)血液中游离DNA的总量很低,每10ml全血平均只能提取到50ng左右的游离DNA,而肿瘤相关突变种类很多,常规技术无法使用这么少的DNA进行多项DNA突变检测实验。对于多重基因的检测,目前最常使用的技术是二代测序(NGS),以Illumina公司的Hiseq和Nextseq测序仪为代表的第二代测序技术,具有检测通量高(每次运行可以产生上百G的基因数据),覆盖面广(可以进行人类全基因组的测序分析),性价比高(平均每G成本仅需几十块钱)等优点,因此特别适用于多个基因的多种突变类型的并行检测。在进行二代测序文库构建的过程中,无可避免地会进行PCR反应,而PCR的过程势必会引入碱基的错误。据报道,一般的高保真酶的复制错误率在10-6左右,并且会随着PCR循环数的增多,这个数值还会增大;另外,目前测序精度最高的测序仪Hiseq和Nextseq的单碱基测序错误率都在0.01%-1%之间。以上这两点对于检测超低频的ctDNA突变会造成很大的背景噪音。在0.1%及以下的检测限的情况下,很难区分模板DNA的突变和PCR及测序的错误,这样会导致检测的特异度降低。为了解决ctDNA的超低频检测背景噪音高的问题,目前常规的办法为提高测序深度,通过加大测序的数据量来增加变异检测时的reads(读段)支持数,从而排除测序错误。但是,由于ctDNA的模板含量低,大数据量会造成大量的复制品(duplicate),测序深度的增加与数据量的增加并不是具有线性关系,因此,在造成数据浪费的同时还会由于测序深度的限制导致检测限并不是很高,通常只能检测0.5%左右的突变。另一种办法为通过模板DNA分子的内源性“标签”,即DNA分子两端的碱基序列来判定下机数据中的reads是否来自于同一条DNA原始分子,来源于同一条DNA原始分子的reads将会根据每条reads的突变情况确定真实的突变情况。但是,由于内源性“标签”所标记的DNA分子数有限,会出现多个DNA原始分子标签序列相同的情况,这样会导致检测的灵敏度下降。技术实现要素:本发明旨在提供一种ctDNA超低频突变检测文库的构建方法、试剂盒及文库检测数据的分析方法,以提高ctDNA超低频突变检测灵敏度。为了实现上述目的,根据本发明的一个方面,提供了一种ctDNA超低频突变检测文库的构建方法。该构建方法包括以下步骤:S1,从全血中提取cfDNA;S2,对cfDNA进行末端修复及3’端添加A碱基;S3,将S2得到的cfDNA的末端连接含有随机标签序列的接头;S4,根据接头的序列及目标区域设计多重PCR的引物进行目标区域捕获,多重PCR的引物中上游引物为通用引物,匹配接头的序列,下游引物为扩增目标区域的特异性引物;S5,对S4的PCR产物进行磁珠纯化,去除掉未非特异性扩增的小片段DNA及引物二聚体;以及S6,对S5的产物进行PCR扩增,同时引入index序列,得到ctDNA超低频突变的文库。进一步地,随机标签序列的长度为12bp。进一步地,接头包括如SEQIDNO.1和SEQIDNO.2所示的脱氧核苷酸序列。根据本发明的另一方面,提供了一种用于ctDNA超低频突变检测文库构建的试剂盒。该试剂盒包括:cfDNA提取试剂、对cfDNA进行末端修复及3’端添加A碱基的试剂、含有随机标签序列的接头、接头连接试剂、根据接头的序列及目标区域设计的多重PCR引物,多重PCR引物中上游引物为通用引物,匹配接头的序列,下游引物为扩增目标区域的特异性引物,对PCR产物进行磁珠纯化的试剂,去除掉未非特异性扩增的小片段DNA及引物二聚体的试剂,用于PCR扩增的试剂,将index序列引入DNA片段的引物。进一步地,随机标签序列的长度为12bp。进一步地,接头包括如SEQIDNO.1和SEQIDNO.2所示的脱氧核苷酸序列。根据本发明的再一方面,提供了一种文库检测数据的分析方法。该分析方法中文库上述ctDNA超低频突变检测文库,包括以下步骤:S1,对文库测序后的数据通过随机标签序列进行聚类分析,排除掉PCR扩增错误及测序错误产生的序列数据;S2,剩余的数据质控合格后进入变异检测分析。进一步地,S2包括:采用samtools软件进行突变检测,并根据聚类完的标签序列进行计数,在同一标签中支持突变的reads占80%以上则计数,所有标签在同一个突变出现2次算作阳性检出,合并点突变检出结果,使用annovar软件进行注释,得到氨基酸水平突变注释。应用本发明的技术方案,通过对二代测序前文库中的每一条目标序列添加外源的“标签”,文库测序后的数据可通过标签序列进行聚类分析,识别出原始的DNA模板,排除掉PCR错误及测序错误,提高检测的特异度。并且,由于添加的是外源标签,可以通过增加标签的多样性来防止多个DNA分子添加相同标签序列的情况,从而保证检测的灵敏度不受影响。附图说明构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:图1示出了根据本发明实施方式的ctDNA超低频突变检测的流程示意图;图2示出了根据本发明实施方式的ctDNA超低频突变检测的文库的构建方法的原理示意图;以及图3示出了实施例1中未加随机标签序列的测序结果在基因组目标区域上每个位点的背景噪音图;以及图4示出了实施例1中加随机标签序列以后的测序结果在基因组目标区域上每个位点的背景噪音图。具体实施方式需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。根据本发明一种典型的实施方式,提供一种ctDNA超低频突变检测文库的构建方法。该构建方法包括以下步骤:S1,从全血中提取cfDNA(cellfreeDNA);S2,对cfDNA进行末端修复及3’端添加A碱基;S3,将S2得到的cfDNA的末端连接含有随机标签序列的接头;S4,根据接头的序列及目标区域设计多重PCR的引物进行目标区域捕获,多重PCR的引物中上游引物为通用引物,匹配接头的序列,下游引物为扩增目标区域的特异性引物;S5,对S4的PCR产物进行磁珠纯化,去除掉未非特异性扩增的小片段DNA及引物二聚体;以及S6,对S5的产物进行PCR扩增,同时引入index序列,得到ctDNA超低频突变的文库。应用本发明的技术方案,通过对二代测序前文库中的每一条目标序列添加外源的“标签”,文库测序后的数据可通过标签序列进行聚类分析,识别出原始的DNA模板,排除掉PCR错误及测序错误,提高检测的特异度。并且,由于添加的是外源标签,可以通过增加标签的多样性来防止多个DNA分子添加相同标签序列的情况,从而保证检测的灵敏度不受影响。本发明中,通过对目标DNA分子添加随机标签来进行标记;添加随机标签的建库方法是基于多重PCR目标区域捕获技术,采用通用的上游引物及目标区域特异性的下游引物来实现文库构建的;通过对添加的随机标签序列进行聚类分析,通过分子计数来区分真突变及PCR或测序错误。优选的,随机标签序列为ATGC四种碱基组成,长度为12bp,分别连接在cfDNA的前后两端。根据本发明一种典型的实施方式,接头包括如SEQIDNO.1和SEQIDNO.2所示的脱氧核苷酸序列。根据本发明一种典型的实施方式,提供一种用于ctDNA超低频突变检测文库构建的试剂盒。该试剂盒包括:cfDNA提取试剂、对cfDNA进行末端修复及3’端添加A碱基的试剂、含有随机标签序列的接头、接头连接试剂、根据接头的序列及目标区域设计的多重PCR引物,多重PCR引物中上游引物为通用引物,匹配接头的序列,下游引物为扩增目标区域的特异性引物,对PCR产物进行磁珠纯化的试剂,去除掉未非特异性扩增的小片段DNA及引物二聚体的试剂,用于PCR扩增的试剂,将index序列引入DNA片段的引物。优选的,随机标签序列为ATGC四种碱基组成,长度12bp,分别连接在readscfDNA的前后两端。根据本发明一种典型的实施方式,接头包括如SEQIDNO.1和SEQIDNO.2所示的脱氧核苷酸序列。根据本发明一种典型的实施方式,提供一种文库检测数据的分析方法,文库为上述ctDNA超低频突变检测文库。该分析方法包括以下步骤:S1,对文库测序后的数据通过随机标签序列进行聚类分析,排除掉PCR扩增错误及测序错误产生的序列数据;S2,剩余的数据质控合格后进入变异检测分析。优选的,S2包括:采用samtools软件进行突变检测,并根据聚类完的标签序列进行计数,在同一标签中支持突变的reads占80%以上则计数,所有标签在同一个突变出现2次算作阳性检出,合并点突变检出结果,使用annovar软件进行注释,得到氨基酸水平突变注释。本发明涉及了一种针对血液ctDNA样本进行目标区域捕获的超低频检测的二代测序文库的构建及数据分析方法。该方法包括了设计特定的测序接头序列,接头序列尾端加上一定数量的随机碱基(即上文所述“标签”);目标区域及标签的扩增,测序接头的引入、Illumina平台HiseqX测序以及自主开发的针对此方法的信息分析流程等步骤。本发明提供了一种利用NGS技术检测血浆ctDNA超低频突变的文库构建方法及数据分析方法。采用本发明提供的方法,可以有效的去除掉由于PCR及测序引入的错误突变,降低检测的背景噪音。对于0.1%的检测限,其灵敏度可以达到90%以上,特异度高达99.9%。如图1所示,应用本发明的技术方案主要包括实验流程和信息分析流程两大部分,实施上述技术方案包括具体以下步骤(图1示出了最主要的步骤):1)从全血中分离血浆,并从血浆中提取cfDNA;2)含有随机标签序列的接头制作;3)cfDNA片段末端修复及3’端添加A碱基;4)接头的连接,将末端连接A碱基的DNA片段末端连接步骤2)中的含有随机标签序列的接头;5)基于多重PCR的目标区域捕获:根据步骤4)中添加的接头序列及目标区域(感兴趣的研究位点),设计多重PCR的引物,其中上游引物为通用引物,匹配步骤4)中添加的接头序列;下游引物为目标区域特异性引物;6)对步骤5)中的PCR产物进行磁珠纯化,去除掉未非特异性的小片段DNA及引物二聚体;7)文库的扩增及index的引入:采用特定的引物对步骤6)后的产物进行PCR扩增,同时对文库添加index序列;8)文库检测:使用安捷伦2100Bioanalyzer检测插入片段大小;使用qPCR定量检测文库产量;9)文库上机:采用IlluminaHiseqX平台对文库检测后的文库进行上机测序;10)下机数据的分析。本发明的ctDNA超低频突变检测的文库的构建方法的原理示意图如图2所示。其中第一步为模板DNA分子(1为待测位点)与含随机标签序列的接头连接(2为随机标签序列);第二步为多重PCR扩增目标区域(3为通用序列的上游引物,4为含有一段通用序列的下游引物);第三步为文库富集及引入测序接头和index序列(5为index序列);第四为构建后的文库结构的示意图(6和7均为测序接头)。下面将结合实例进一步说明本发明的有益效果。实施例11.非小细胞肺癌患者全血10ml,采用streck公司的BCT管进行收集和运输,运输温度为室温,运输时间不超过72h。血浆的分离采用两步离心法,即1600g离心10min,取上清,再16000g离心10min,上清即为分离好的血浆,该血浆保存于-80℃中。血浆中cfDNA的提取采用Qiagen公司的circulating循环DNA提取试剂盒,提取好的cfDNA存放在-20℃中备用。样本名称及提取量见表1。表12.合成带有标签序列的接头。接头序列见表2(本例中采用的标签序列长度为12个碱基),最多可以标记412个DNA分子。合成好的引物采用EB(ElutionBuffer)洗脱缓冲液进行溶解,终浓度为100uM,等比例摩尔数混合之后95℃加热5min,然后缓慢降温至室温完成退火。采用乙醇沉淀法对退火完成后的接头进行纯化,最后采用100ul无核酸酶水溶解,终浓度为20uM。表2名称序列(5’-3’)SEQIDNO.1TCTACACTCTTTCCCTACACGCTCTTCCGATCTNNNNNNNNNNNNTSEQIDNO.2CACTGACCTCAAGTCTGCACACGAGAAGGCTAGANNNNNNNNNNNN注:含有标签序列的接头,其中N代表随机碱基。3.末端修复及3’端加A碱基参照下表3配比准备反应混合液,用枪轻柔地上下吹吸混匀。表3放入PCR仪,按下表4设置程序进行反应(盖温设为70℃,前30min不盖热盖,温度到达65℃,立即盖上热盖)。表44.接头连接按照下表5的配比准备反应混合液,用枪轻柔地上下吹吸混匀。表5分为2管,每管55ul,放于PCR仪上,20℃反应15min。反应结束后,使用0.8×的AMPureXP(Beckman公司)磁珠纯化DNA样本。5.PCR扩增捕获目标区域按照下表6的配比准备反应混合液,用枪轻柔地上下吹吸混匀。表6用于多重PCR扩增的上下游引物的序列见表7。表7名称序列(5’-3’)SEQIDNO.3TCTACACTCTTTCCCTACACGACGCTSEQIDNO.4GGAGTTCAGACCGTGTGCTCTTCCGATCTN注:下游引物为混合物,N代表针对不同的位点设计的不同特异性引物,具体的N部分序列见表8。表8放入PCR仪,按下表9设置程序反应。表9反应结束后,采用1.2×的AMPureXP磁珠对扩增产物进行纯化,最后将文库溶于25ulNF-water(Ambion公司)中。6.文库的富集及引入index序列采用含有Illumina的index引物对上一步的文库进行扩增,同时引入index序列在上机时区分文库,在本实施例中,采用的index序列为1号,按照下表10配制反应液。表10上下游引物序列见表11。表11引物名称序列(5’-3’)SEQIDNO.48AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACASEQIDNO.49CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCA放入PCR仪,按下表12设置程序反应。表12反应结束后,采用1.2×的AMPureXP磁珠对扩增产物进行纯化,最后将文库溶于30ulNF-water中。采用Qubit荧光计对文库进行精确定量。7.文库检测及上机测序将步骤6中的纯化产物稀释到2ng/ul,取出1ul进行安捷伦2100Bioanalyzer(美国安捷伦公司)检测;另外,再取出1ul用于qPCR检测,根据检测结果决定上机浓度。根据上步所得的浓度,将文库稀释到上机要求后(2nmol),在Illumina公司的HiseqX测序平台上进行PE150测序。8.下机质控要求总数据量大于2G,目标区域数据占比大于80%,平均测序深度50000X以上。合并相同标签序列时平均同一标签支持数8个reads以上。合并后平均测序深度3000X以上,所有目标区域测序深度大于2000X的在90%以上,实施例中的样本质控数据见表13。表139.突变分析质控合格的数据进入变异检测分析,对做好标记的序列采用samtools软件进行突变的检测,并且根据聚类好的标签序列进行计数,在同一标签中支持突变的reads占80%以上就计数。所有标签在同一个突变出现2次就算作阳性检出。合并点突变检出结果,并且使用annovar软件进行注释,得到氨基酸水平突变注释。对比数据库找到癌症靶向用药相关检测结果。本实施例中的检测结果,WHX样本的突变位点检出情况见表14。表1410.数字PCR对检测结果的验证对于检测到的低频位点,EGFRp.T790M,为了证明该方法检测的准确性,采用目前公认的灵敏度最高的检测方法,数字PCR来进行验证。验证的结果为带有0.15%频率的EGFR:p.T790M阳性突变,这与本发明提供的方法的检测结果十分一致。图3和图4所示为采用实施例1中的方法未加随机标签序列和添加标签序列对同一个正常人的cfDNA样本进行检测的背景噪音图。在正常人的样本中,我们默认是不会有很多0.5%以下的突变。如图3所示,对于未加随机标签序列的实验而言,检测到非常多的0.5%突变频率以下的突变位点。而添加标签序列后,如图4所示,0.2%突变频率的位点仅有少数。从单核苷酸突变频率上来看,未加随机标签序列的实验,单核苷酸的突变率达到了2.7*10-4,本发明提供的方法单核苷酸的突变率仅有4.3*10-6,更加接近文章中报道的正常人的突变率(3.0*10-6)。现有的技术对于检测超低频(突变频率0.5%以下)突变没有十分好的办法,通常会采用加大测序数据量,从而增加突变的reads支持数来判定真阳性和假阳性,由于ctDNA的含量极低,这种方式会造成数据量的大量浪费,同时对于频率在0.5%以下的突变位点并不能很好的进行区分。另一种方法是根据分子内源性“标签”来校正背景噪音,但是由于内源性的分子“标签”有限,导致假阴性率较高。本发明提供的方法通过添加外源性的分子“标签”,经过聚类分析排除掉PCR引入的错误以及测序错误,从而有效地去除掉背景噪音,使得检测0.1%左右的突变的准确性大大提高。以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。SEQUENCELISTING<110>天津诺禾医学检验所有限公司<120>ctDNA超低频突变检测文库的构建方法、试剂盒及文库检测数据的分析方法<130>PN58854NHZY<160>49<170>PatentInversion3.5<210>1<211>46<212>DNA<213>artificial<220><223>含有随机标签序列的接头<220><221>misc_feature<222>(34)..(45)<223>nisa,c,g,ort<400>1tctacactctttccctacacgctcttccgatctnnnnnnnnnnnnt46<210>2<211>46<212>DNA<213>artificial<220><223>含有随机标签序列的接头<220><221>misc_feature<222>(35)..(46)<223>nisa,c,g,ort<400>2cactgacctcaagtctgcacacgagaaggctagannnnnnnnnnnn46<210>3<211>26<212>DNA<213>artificial<220><223>用于多重PCR扩增的上游引物<400>3tctacactctttccctacacgacgct26<210>4<211>30<212>DNA<213>artificial<220><223>用于多重PCR扩增的下游引物<220><221>misc_feature<223>N代表针对不同的位点设计的不同特异性引物<220><221>misc_feature<222>(30)..(30)<223>nisa,c,g,ort<400>4ggagttcagaccgtgtgctcttccgatctn30<210>5<211>24<212>DNA<213>artificial<220><223>NRAS_1R<400>5cacccccaggattcttacagaaaa24<210>6<211>23<212>DNA<213>artificial<220><223>NRAS_2R<400>6caagtgtgatttgccaacaagga23<210>7<211>24<212>DNA<213>artificial<220><223>NRAS_3R<400>7gttcttgctggtgtgaaatgactg24<210>8<211>26<212>DNA<213>artificial<220><223>PIK3CA_1R<400>8agaaaaccattacttgtccatcgtct26<210>9<211>25<212>DNA<213>artificial<220><223>PIK3CA_2R<400>9gcacttacctgtgactccatagaaa25<210>10<211>27<212>DNA<213>artificial<220><223>PIK3CA_3R<400>10cataagagagaaggtttgactgccata27<210>11<211>27<212>DNA<213>artificial<220><223>PIK3CA_4R<400>11caaacaagtttatatttccccatgcca27<210>12<211>24<212>DNA<213>artificial<220><223>PIK3CA_5R<400>12tgctgttcatggattgtgcaattc24<210>13<211>28<212>DNA<213>PIK3CA_6R<400>13agcatcagcatttgactttaccttatca28<210>14<211>28<212>DNA<213>artificial<220><223>PIK3CA_7R<400>14gtggaagatccaatccatttttgttgtc28<210>15<211>22<212>DNA<213>artificial<220><223>PIK3CA_8R<400>15ggttgaaaaagccgaaggtcac22<210>16<211>33<212>DNA<213>artificial<220><223>PIK3CA_9R<400>16tttaagattacgaaggtattggtttagacagaa33<210>17<211>28<212>DNA<213>artificial<220><223>PIK3CA_10R<400>17tcaatcagcggtataatcaggagttttt28<210>18<211>28<212>DNA<213>artificial<220><223>PIK3CA_11R<400>18ccttttgtgtttcatccttcttctcctg28<210>19<211>28<212>DNA<213>artificial<220><223>EGFR_1R<400>19tcagtccggttttatttgcatcatagtt28<210>20<211>23<212>DNA<213>artificial<220><223>EGFR_2R<400>20gtgccagggaccttaccttatac23<210>21<211>22<212>DNA<213>artificial<220><223>EGFR_3R<400>21tccagaccagggtgttgttttc22<210>22<211>20<212>DNA<213>artificial<220><223>EGFR_4R<400>22cggacatagtccaggaggca20<210>23<211>21<212>DNA<213>artificial<220><223>EGFR_5R<400>23ccccatggcaaactcttgcta21<210>24<211>26<212>DNA<213>artificial<220><223>EGFR_6R<400>24gcatgtgttaaacaatacagctagtg26<210>25<211>20<212>DNA<213>artificial<220><223>EGFR_7R<400>25ctgaggttcagagccatgga20<210>26<211>25<212>DNA<213>artificial<220><223>EGFR_8R<400>26cccaaagactctccaagatgggata25<210>27<211>23<212>DNA<213>artificial<220><223>MET_1R<400>27agaagttgatgaaccggtccttt23<210>28<211>27<212>DNA<213>artificial<220><223>MET_2R<400>28tctgacttggtggtaaacttttgagtt27<210>29<211>27<212>DNA<213>artificial<220><223>MET_3R<400>29gcaaaccacaaaagtatactccatggt27<210>30<211>27<212>DNA<213>artificial<220><223>MET_4R<400>30ggagacatctcacattgtttttgttga27<210>31<211>29<212>DNA<213>artificial<220><223>MET_5R<400>31cggtagtctacagattcatttgaaaccat29<210>32<211>27<212>DNA<213>artificial<220><223>MET_6R<400>32gcttttcaaaaggcttaaacacaggat27<210>33<211>23<212>DNA<213>artificial<220><223>MET_7R<400>33aggccccatacaatttgatgaca23<210>34<211>21<212>DNA<213>artificial<220><223>RET_1R<400>34ccttgttgggacctcagatgt21<210>35<211>26<212>DNA<213>artificial<220><223>RET_2R<400>35actttgcgtggtgtagatatgatcaa26<210>36<211>21<212>DNA<213>artificial<220><223>RET_3R<400>36gtggtagcagtggatgcagaa21<210>37<211>18<212>DNA<213>artificial<220><223>RET_4R<400>37ccatggtgcacctgggat18<210>38<211>22<212>DNA<213>artificial<220><223>ERBB2_1R<400>38gccatagggcataagctgtgtc22<210>39<211>23<212>DNA<213>artificial<220><223>ERBB2_2R<400>39ccttggtccttcacctaaccttg23<210>40<211>23<212>DNA<213>artificial<220><223>ERBB2_3R<400>40gtcatatctccccaaaccccaat23<210>41<211>21<212>DNA<213>artificial<220><223>ALK_1R<400>41ggaagagtggccaagattgga21<210>42<211>22<212>DNA<213>artificial<220><223>ALK_2R<400>42gcccagactcagctcagttaat22<210>43<211>29<212>DNA<213>artificial<220><223>KRAS_1R<400>43gtaaaaggtgcactgtaataatccagact29<210>44<211>27<212>DNA<213>artificial<220><223>KRAS_2R<400>44gactctgaagatgtacctatggtccta27<210>45<211>27<212>DNA<213>artificial<220><223>KRAS_3R<400>45aggcctgctgaaaatgactgaatataa27<210>46<211>27<212>DNA<213>artificial<220><223>BRAF_1R<400>46tttctttttctgtttggcttgacttga27<210>47<211>28<212>DNA<213>artificial<220><223>BRAF_2R<400>47gcttgctctgataggaaaatgagatcta28<210>48<211>41<212>DNA<213>artificial<220><223>引入index序列的上游引物<400>48aatgatacggcgaccaccgagatctacactctttccctaca41<210>49<211>44<212>DNA<213>artificial<220><223>引入index序列的下游引物<400>49caagcagaagacggcatacgagatcgtgatgtgactggagttca44当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1