一种RFFT1细胞的构建方法与流程
本发明涉及生物技术领域,尤其涉及一种rfft1细胞的构建方法。
背景技术:
肿瘤细胞免疫治疗是一种新兴的肿瘤治疗模式,它从病人体内采集免疫细胞,然后进行体外培养和扩增,再回输到病人体内,来激发以及增强机体的自身免疫功能以治疗肿瘤。肿瘤细胞免疫治疗是继手术、放疗和化疗之后的第四种肿瘤治疗方法。
正常或生物工程改造过的人体细胞移植或输入患者体内,新输入的细胞可以替代受损细胞、或者具有更强的免疫杀伤功能,从而达到治疗疾病的目的。
生物工程改造过的细胞用特殊方法在体外培养过程中加以改造,可有效杀除患者体内肿瘤细胞。如,中国专利申请cn201210194280.5提供了一种人细胞因子诱导的杀伤细胞。中国专利申请cn201510034781.0提供一种肿瘤细胞特异性多克隆t细胞。中国专利申请cn201510013987.5提供一种抗肿瘤t细胞及其制备方法。中国专利申请cn201711060030.1提供一种治疗艾滋病合并淋巴瘤的car-t细胞及其制备方法和应用。cn201610824893.0提供一种抗体调控的双抗原特异性t细胞及其制备方法和应用。
现有技术中,一般是通过dc细胞递呈t细胞,产生特异杀伤的t细胞,或者用病毒做为载体,通过慢病毒转染技术诱导t细胞的特异性杀伤。但要么由于肿瘤抗原不明和肿瘤微环境免疫抑制的障碍,致使效果不佳;要么由于让较为单薄的特异性细胞直接面对复杂的肿瘤微环境,因而导致效果不佳,要么靶点单一,只对个别肿瘤有效。
技术实现要素:
本发明旨在提供一种rfft1细胞的构建方法,以构建一种新的tcr-t细胞(命名为rfft1细胞),用于细胞免疫治疗,实现了查杀的精准性、特异性和安全性。
本发明提供一种rfft1细胞的构建方法,所述构建方法包括以下步骤:
s1)对肿瘤细胞进行全外显子测序;将全外显子测序结果与正常细胞的基因组相比,筛选出突变的氨基酸位点;以突变的氨基酸位点为中心预测抗原表位;采用多肽固相合成法合成突变多肽;pbmc细胞负载致肿瘤突变的多肽,而后对负载多肽后的pbmc细胞进行一次多肽冲击;
s2)冲击后扩大培养,得到ff细胞;
s3)以致肿瘤突变的多肽作为抗原直接刺激所述ff细胞以筛选精准多肽;
s4)培养pbmc细胞,在培养过程中,用精准多肽进行多次多肽冲击;
s5)冲击后继续培养,得到rff细胞;
s6)以精准多肽作为抗原刺激所得rff细胞,筛选获得能够识别所述精准多肽的特异性细胞;
s7)通过测序得到特异性细胞的tcrβ链cdr3区序列,由所述tcrβ链cdr3区序列扩增得到tcr基因;
s8)培养pbmc细胞,敲除细胞中原有的tcr基因和细胞表面免疫抑制性信号分子,并转入步骤s7中获得的能够与精准多肽特异性结合的tcr基因,制备得到rfft1细胞,所述免疫抑制性信号分子包括:pd-1、tim-3、lag3、ctla-4、btla、vista、cd160、tigit、2b4(cd244)。
进一步地,所述肿瘤细胞来源于市售工程细胞系,包括h1299、h226、h358、h1563、h2228、a549、renca、llc小鼠lewis肺癌细胞、crl-6323b16f1、crl-25394t1、u14小鼠子宫颈癌细胞、bv-2小鼠小胶质瘤细胞、g422小鼠神经胶质瘤细胞。
进一步地,所述抗原表位的预测是以突变的氨基酸位点为中心,向两侧延伸10个氨基酸,以一段21个氨基酸的多肽作为潜在抗原表位作为潜在抗原表位。
进一步地,在步骤s2中,所述冲击后扩大培养,得到ff细胞组合物包括:
将多肽冲击后的pbmc细胞在预铺细胞刺激因子okm-25的细胞培养装置中培养5天;
转移至含有培养液okm-100+12%fbs的细胞培养装置中继续培养至第10天;
转移至含有培养液okm-200+5%fbs的细胞培养装置中继续培养至第14~21天。
进一步地,步骤s4中,重复多肽冲击3~4次。
进一步地,多肽冲击中,用浓度为10μg/ml~100μg/ml的多肽溶液进行多肽冲击。
进一步地,多肽冲击的冲击时间1-4h。
步骤s8中,用crispr技术依次敲除细胞免疫抑制性信号分子以及细胞原有tcr基因。
步骤s8中,用敲除原有tcr的crispr慢病毒载体和敲除表面免疫抑制性信号分子的crispr慢病毒载体去感染pbmc细胞,同时进行原有tcr的敲除以及免疫抑制性信号分子的去除;而后转入表达步骤s7所获得tcr基因的tcr基因表达载体,以转入步骤s7所获得的tcr基因。
本发明具有如下有益效果:
1)本发明提供一种新的细胞构建方法,用于构建一种新的tcr-t细胞(命名为rfft1细胞),是一种攻防兼备的超级t细胞。在该方法中,通过联合混合多肽技术、精准多肽二次冲击、tcr-t技术、靶点敲除防护技术,对t细胞进行改造。首先进行混合多肽冲击刺激,而后筛选出有效的精准多肽以其对pbmc细胞进行再次多肽冲击,且进行加量的多肽冲击刺激。在两次刺激的基础上,又联合tcr-t技术进行第三次刺激,进而将普通t细胞改造为更有特异杀伤作用的t细胞,杀伤效率高、精准性高。
2)在本发明中,同时联合靶点敲除防护技术、体外培养和体外扩增实现了查杀的精准性、特异性和安全性,覆盖更多瘤种,提高具有高精准性杀伤作用的t细胞对复杂的肿瘤微环境的适应力。
3)与直接用慢病毒转染诱导t细胞的杀伤作用相比,简单方便、安全性高。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1示出了抗原负载效率检测;
图2示出了精准多肽筛选结果;
图3示出了流式检测特异性t细胞比例;
图4示出了特异性细胞的tcr分布情况;
图5示出了流式检测细胞上原有tcr的敲除情况;
图6示出了免疫抑制性信号分子体外封闭情况;
图7示出了构建tcr的表达情况;
图8示出了本发明实施例所述rfft1细胞对靶细胞的杀伤作用;
图9示出了本发明实施例所述rfft1细胞释放细胞因子的检测;
图10示出了荷瘤小鼠生存曲线。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
肿瘤细胞免疫治疗是一种新兴的肿瘤治疗模式。在肿瘤细胞免疫治疗方面,现有的lak、dc、cik、dc-cik细胞已基本被证明无效。本发明提供一种新的t细胞,可用于细胞免疫治疗。本发明对t细胞进行改造,提供一种tcr-t细胞,杀伤力强、精准性高、覆盖多种瘤种。
本发明实施例提供rfft1细胞的构建方法,构建了一种用于细胞免疫治疗的tcr-t细胞(本文中命名为rfft1细胞)。该构建方法可以包括以下步骤:
s1)对肿瘤细胞进行全外显子测序;将全外显子测序结果与正常细胞的基因组相比,筛选出突变的氨基酸位点;以突变的氨基酸位点为中心预测抗原表位;采用多肽固相合成法合成突变多肽;pbmc细胞负载致肿瘤突变的多肽,而后对负载多肽后的pbmc细胞进行多肽冲击;
s2)冲击后扩大培养,得到ff细胞;
s3)以致肿瘤突变的多肽作为抗原直接刺激所述ff细胞以筛选精准多肽;
s4)培养pbmc细胞,在培养过程中,用精准多肽进行多肽冲击;
s5)冲击后继续培养,得到rff细胞;
s6)以精准多肽作为抗原刺激所得rff细胞,筛选获得能够识别所述精准多肽的特异性细胞;
s7)通过测序得到特异性细胞的tcrβ链cdr3区序列,由所述tcrβ链cdr3区序列扩增得到tcr基因;
s8)敲除t细胞中原有的tcr基因和表面免疫抑制性信号分子,并转入步骤s7中获得的能够与精准多肽特异性结合的tcr基因,制备得到rfft1细胞,所述免疫抑制性信号分子包括:pd-1、tim-3、lag3、ctla-4、btla、vista、cd160、tigit、2b4(cd244)。
本文中,定义“rfft1”细胞涉及的含义如下:
r—精准多肽二次冲击技术;
ff—混合多肽技术;
t—tcr-t技术;
1—靶点敲除防护技术。
即,本文中“rfft1”细胞为通过联合混合多肽技术、精准多肽二次冲击技术、tcr-t(t细胞受体(tcr)嵌合型t细胞)技术、靶点敲除防护技术所组成的rfft1方案制备得到的一种新的t细胞,可用于肿瘤免疫治疗。顾名思义,“ff”细胞为由ff方案得到的t细胞。“rff”细胞为由rff方案得到的t细胞。“rfft”细胞为由rfft方案得到的t细胞。
本发明实施例提供了一种用于细胞免疫治疗的tcr-t细胞。本发明实施例的rfft1方案中,首先,用混合多肽直接对负载多肽后的pbmc细胞进行冲击,进行第一次刺激;其次,筛选精准多肽并用筛选得到的精准多肽作为抗原直接刺激ff细胞,进行第二次刺激;随后通过tcr-t技术进行第三次刺激。通过三次刺激对t细胞进行了改造,改造后的t细胞实现了查杀的精准性、特异性和安全性。同时,结合体外培养和靶点敲除防护技术,使改造后的t细胞提高了对体内复杂免疫环境的适应力。
在步骤s1中,所述致肿瘤突变的多肽可以由以下方法合成得到:1)外显子测序;2)抗原表位预测;3)合成多肽。
1)外显子测序
对肿瘤细胞进行全外显子测序,而后利用软件对测序信息进行分析,一方面得到mhc类型信息;另一方面,将全外显子测序结果与正常细胞的基因组相比,筛选出突变的氨基酸位点。
在外显子测序中,肿瘤细胞可以来自于工程细胞系,也可以来自于患者外周血。
工程细胞系可以包括h1299、h226、h358、h1563、h2228、a549、renca、llc小鼠lewis肺癌细胞、crl-6323b16f1、crl-25394t1、u14小鼠子宫颈癌细胞、bv-2小鼠小胶质瘤细胞、g422小鼠神经胶质瘤细胞。工程细胞系是指采用基因工程技术或细胞融合技术对宿主细胞的遗传物质进行修饰改造或重组,获得具有稳定遗传的独特性状的细胞系。
2)抗原表位预测
在抗原表位预测中,以突变的氨基酸位点为中心,向两侧延伸10个氨基酸,将这段21个氨基酸的多肽作为“潜在抗原表位”。使用预测软件分析潜在抗原表位的ic50(推荐软件:netmhcpan3.0、pickpocket、artificialneuralnetworks(ann))。若ic50<1000nm则将此潜在抗原表位认定为“抗原表位”。
3)合成多肽
采用多肽固相合成法合成抗原表位肽。
具体地,pbmc细胞负载致肿瘤突变的多肽,而后对负载多肽后的pbmc细胞进行多肽冲击具体可以为:
1)配制多肽溶液:以rpmi1640+10%fbs(胎牛血清)或okm100+12%fbs溶解多肽,多肽的终浓度为10~100μg/ml,优选50μg/ml,备用;
2)pbmc提前1天复苏,吹打细胞,吸取15ml,并计数,离心;
3)以配制的多肽溶液将pbmc重悬;
4)置于细胞培养板中进行冲击;
5)37℃5%co2,冲击1~4h,优选4h,得到冲击后的pbmc细胞,备用。
在步骤s2中,冲击后扩大培养以得到ff细胞具体可以为:
1)刺激因子okm-25预铺板,40μlokm-25+4mlpbs(磷酸缓冲盐溶液)、2ml/皿(4.5cm2)室温4h,4℃备用;
2)将冲击后的pbmc转移至预铺omk-25的细胞培养板或培养瓶中;
3)晃匀,37℃5%co2培养,记为第0天;
4)观察共培养细胞情况,根据细胞密度,在第5天,将共培养细胞转移至大培养瓶中,补新鲜的培养液okm-100+12%fbs,20ml于75cm2培养瓶中;
5)共培养第7天,再加入20ml新鲜okm-100+12%fbs;
6)共培养第10天,培养液为okm-200+5%fbs,将共培养细胞从75cm2瓶中转移至175cm2大瓶中;
7)25ml培养液okm-200+5%fbs吹打,再转入大瓶,重复2次;以培养液okm-200+5%fbs补足至200ml。
8)培养至14-21天时,可获得ff细胞组合物。通过离心从ff细胞组合物中提取t细胞,即为ff细胞。
步骤s3中,从所述ff细胞组合物中分离提取t细胞,以致肿瘤突变的多肽作为抗原直接刺激所述t细胞以筛选精准多肽。
精准多肽的筛选标准为:
多肽作为抗原的以ff细胞作为基线;两个独立重复,检测值高的为高基线,低的为低基线;
两个基线的差异为系统误差,数据分析时,对检测值>低基线、>高基线以及>高基线+系统误差的,分别进行标注;检测值>高基线+系统误差即为有效的精准多肽。
步骤s3中,以多肽作为抗原直接刺激t细胞以筛选精准多肽具体可以为:
1)离心所获得的ff细胞组合物,1500rpm离心5min收集t细胞,加入10mlpbs重悬细胞并计数,1500rpm离心5min,收集t细胞以1640+10%fbs+200u/mlil2重悬,计数调整至1×106个/ml;
2)以排枪将t细胞分至96孔平底板中,200μl/孔,细胞数为2×105个;再分别加入1mg/ml的突变多肽10μl,终浓度为50μg/ml,每条多肽设置3复孔;
3)设置阳性对照:t细胞+100ng/mlokt3(cd3单克隆抗体);阴性对照:rpmi1640+10%fbs+200u/mlil2(白细胞介素2);两个t细胞对照作为本底释放检测,分别为第一次加t细胞,和最后一次加t细胞;以两个本底释放的差异作为系统误差;
4)37℃、5%co2刺激24h后,1500rpm离心10min,转移140μl上清至新的96孔板中;
5)再将96孔板进行离心,1500rpm10min,取样品进行elisa(酶联免疫吸附测定)检测(或将样品至于-80℃保存);
6)筛选精准多肽:
基于上述精准多肽筛选标准,检测值>高基线+系统误差即为有效的精准多肽。
步骤s4中,培养pbmc细胞,在培养过程中,用精准多肽进行多肽冲击,进行第二次刺激。
相比步骤s1,在步骤s4中采用加量的精准多肽冲击。也就是说,在该步骤中对pbmc细胞进行多次多肽冲击刺激。具体可以重复多肽冲击3~4次。
培养过程中,培养方案近似于步骤s2中对负载多肽后的pbmc细胞的扩大培养。先在预铺细胞刺激因子okm-25的装置中培养一段时间,而后依次转移至okm-100+12%fbs培养基、okm-200+5%fbs培养基中继续培养。
步骤s4具体可以包括如下步骤:
将pbmc细胞在预铺细胞刺激因子okm-25的细胞培养装置中培养,培养一段时间后以步骤s3所得精准多肽进行多肽冲击;
而后转移至含有培养液okm-100+12%fbs的细胞培养装置(培养板或培养瓶)中继续培养,每隔3~4天分别以步骤s3所得精准多肽进行多肽冲击。
每次多肽冲击的冲击时间可为1-4h。通过加量的精准多肽冲击刺激将普通t细胞改造成为具有更精准的杀伤能力的rff细胞。
步骤s5中,冲击后继续培养,得到rff细胞。适用的培养基可以为okm200+5%fbs。多肽冲击后转移至含有培养液okm200+5%fbs的装置中继续培养,37℃、5%co2下培养即可得到精准多肽二次冲击得到的t细胞,即rff细胞。
步骤s6中,以精准多肽作为抗原刺激所得rff细胞,对刺激后的细胞进行cd8、cd137、ifn-γ的染色,以流式细胞仪进行分选,筛选获得能够识别所述精准多肽的特异性细胞。
步骤s7中,通过测序得到特异性细胞的tcrβ链cdr3区序列。由所述tcrβ链cdr3区序列扩增得到tcr基因。
1)由步骤s6分选得到的细胞进行基因组的提取;
2)对基因组进行tcr测序分析,根据tcr分布频率,确定高频的tcr序列;
3)提取pbmc的mrna,反转录成从dna,根据高频tcr的序列,设计引物,扩增得到tcr基因。
在步骤s8中,可以用crispr技术(clusteredregularlyinterspacedshortpalindromicrepeats,基因编辑技术)敲除细胞原有tcr基因。
步骤s8中,敲除外周血t细胞中原有的tcr基因和表面免疫抑制性信号分子,并转入步骤s7中所得能够与精准多肽特异性结合的tcr基因,以得到rfft1细胞。通过tcr-t技术进行第三次刺激,对t细胞进行改造。
在步骤s8中,可通过crispr慢病毒转染敲除原有的tcr基因和表面免疫抑制性信号分子以及转入步骤s7中所得能够与精准多肽特异性结合的tcr基因。
具体地,分别构建3个慢病毒载体:基于步骤s7所获得tcr基因构建tcr基因表达载体;构建敲除原有tcr的crispr慢病毒载体;构建敲除表面免疫抑制性信号分子的crispr慢病毒载体。然后用所构建的慢病毒载体去感染细胞:将上述所得敲除原有tcr的crispr慢病毒载体和敲除表面免疫抑制性信号分子的crispr慢病毒载体去感染pmbc细胞,敲除原有tcr并敲除免疫抑制性信号分子;而后再用tcr基因表达载体去感染细胞,转入步骤s7所获得tcr基因。感染后继续共培养,即可得到rfft1细胞。
敲除tcr基因的crispr慢病毒载体的构建过程可以为:
1)在pubmed上找到tcr基因的mrna的cds区,并分析tcr的保守区,将保守区进行敲除靶点的预测;
2)设计合成sgrna所需的正向引物和反向引物,将正向引物和反向引物1:1混合后,95℃处理5~60min,再进行缓慢降温,形成sgrna的dna序列;
3)将crispr慢病毒表达载体进行双酶切,并与sgrna对应的双链dna进行连接,转入克隆感受态细胞,12h后,挑取单克隆进行测序,保留测序正确的克隆;
4)提取携带sgrna对应dna序列的crispr慢病毒载体质粒,进行病毒包装。
敲除免疫抑制性信号分子的crispr慢病毒载体的构建可以参照敲除tcr基因的crispr慢病毒载体的构建。
可表达步骤s7所确定的tcr基因的tcr基因表达载体的构建过程为:基于所确定的tcr基因进行病毒包装即可。
本发明实施例提供了一种新的细胞构建方法,以构建一种新的用于肿瘤免疫治疗的t细胞(rfft1细胞),通过联合混合多肽技术、精准多肽二次冲击、tcr-t技术、靶点敲除防护技术,将t细胞改造为精准性好、杀伤性高、可覆盖多种瘤种、对复杂免疫微环境适应力强的tcr-t细胞。
下文进一步对本发明实施例所述的rfft1细胞的构建方法进行详述。
(一)全外显子测序
1)使用工程细胞系进行全外显子测序;
2)利用软件对测序信息进行分析:一方面得到mhc类型信息;另一方面,将全外显子测序结果与正常细胞的基因组相比,筛选出突变位点。
(二)抗原表位预测
1)以突变的氨基酸位点为中心,向两侧延伸10个氨基酸,将这段21个氨基酸的多肽作为“潜在抗原表位”;
2)使用预测软件分析潜在抗原表位的ic50(推荐软件:netmhcpan3.0、pickpocket、artificialneuralnetworks(ann)),如ic50<1000nm则认为此潜在抗原表位为“抗原表位”。
(三)合成多肽
抗原表位肽合成方法采用多肽固相合成法
1)锚定:把第一个氨基酸锚定在固相树脂上;
2)去保护:保护的氨基酸使用碱性溶剂去除氨基的保护基团;
3)活化:使用活化剂活化待连接的氨基酸羧基;
4)键联:活化的羧基与前一个氨基酸裸露的氨基反应,形成肽;
5)重复步骤2-4,使整条抗原表位肽链完全合成。
(四)pbmc负载突变多肽
1)配制多肽溶液:以1640+10%fbs或okm100+12%fbs溶解多肽,每条多肽的终浓度为50μg/ml,备用;
2)pbmc提前1天复苏,吹打细胞,吸取15ml,并计数,离心;
3)以配制的多肽溶液将pbmc重悬,并至于细胞培养板中进行冲击;
4)37℃5%co2,冲击4h,备用。
(五)pbmc经多肽冲击后的扩大培养
1)刺激因子okm-25预铺板,40μlokm-25+4mlpbs,2ml/皿(4.5cm2)室温4h,4℃备用;
2)将冲击后的pbmc转移至预铺omk25的培养瓶中;
3)晃匀,37℃5%co2培养,记为第0天;
4)观察共培养细胞情况,根据细胞密度,在第5天,将细胞转移至大培养瓶中,补新鲜的培养液okm-100+12%fbs,20ml于75cm2培养瓶中;
5)共培养第7天,再加入20ml新鲜okm-100+12%fbs;
6)共培养第10天,培养液为okm-200+5%fbs,将共培养细胞从75cm2瓶中那个转移至175cm2大瓶中;
7)25ml培养液okm-200+5%fbs吹打,再转入大瓶,重复2次;以培养液okm-200+5%fbs补足至200ml。
8)培养至14-21天时,即可获得ff细胞组合物。
(六)多肽作为抗原直接刺激t细胞筛选精准多肽:
1)ff细胞组合物中的t细胞即为ff方案得到的t细胞(ff细胞)。离心收集获得的ff细胞组合物,1500rpm离心5min收集ff细胞,加入10mlpbs重悬细胞并计数,1500rpm离心5min,收集t-cells以1640+10%fbs+200u/mlil2重悬,计数调整至1×106cells/ml;
2)以排枪将ff细胞分至96孔平底板中,200μl/孔,细胞数为2×105cells;再分别加入10μl1mg/ml的突变多肽,终浓度为50μg/ml,每条多肽设置3复孔;
3)设置阳性对照:ff细胞+100ng/mlokt3;阴性对照:1640+10%fbs+200u/mlil2;两个ff细胞对照作为本底释放检测,分别为第一次加ff细胞,和最后一次加ff细胞;以两个本底释放的差异作为系统误差;
4)37℃、5%co2刺激24h后,1500rpm离心10min,转移140μl上清至新的96孔板中;
5)再将96孔板进行离心,1500rpm10min,取样品进行elisa检测(或将样品至于-80℃保存)。
(七)精准多肽评判标准:
1)阳性对照和阴性对照正常,则说明此数据可信;
2)多肽作为抗原的以t-cells作为基线;
3)每组实验为两个基线,高基线和低基线,两个基线的差异为系统误差,数据分析时,对检测值>低基线、>高基线以及>高基线+系统误差的,分别进行标注;检测值>高基线+系统误差即为有效的精准多肽。
(八)以筛选的精准多肽制备rff细胞
1)pbmc以ff细胞组合物培养方案培养(刺激因子okm-25预铺板,40μlokm-25+4mlpbs,2ml/皿(4.5cm2)室温4h,4℃备用;培养条件37℃、5%co2)至第3天,进行精准多肽的再冲击;
2)取2×107的t细胞,加入终浓度为50μg/ml的多肽,冲击4h;
3)冲击4h后,转入25cm2培养瓶,补okm100+12%fbs,37℃5%co2培养,根据细胞生长情况,转移至75cm2培养瓶中,尽量保持细胞密度在1×106个/ml;
4)在培养第7天、第10天和第14天时,重复精准多肽冲击即重复步骤2)和3);
5)细胞进入175cm2培养瓶中时,培养基为okm200+5%fbs,培养10~21天即可得到精准多肽二次冲击得到的t细胞,即rff细胞。
(九)突变抗原特异性杀伤t细胞的培养及分离
1)以精准多肽直接作为抗原对rff细胞进行刺激,刺激12~72h后,备用;
2)对刺激后的细胞进行cd8、cd137、ifn-γ的染色,并以流式细胞仪进行分选,选择cd8+cd137+、或者cd8+ifn-γ+细胞。
(十)cd8+t细胞tcr频率检测及高频tcr的克隆
1)由步骤九分选得到的细胞进行基因组的提取;
2)基因组进行tcr测序分析,根据tcr分布频率,确定高频的tcr序列;
3)提取pbmc的mrna,反转录成从dna,根据高频tcr的序列,设计引物,扩增得到tcr基因;
4)构建tcr基因表达载体,包装病毒。
(十一)构建敲除免疫抑制性信号分子的crispr慢病毒载体
1)免疫抑制性信号分子包括:pd-1、tim-3、lag3、ctla-4、btla、vista、cd160、tigit、2b4(cd244);
2)在pubmed上找到基因的mrna的cds区,分别将每个外显子进行敲除靶点的预测;
3)设计合成sgrna所需的正向引物和反向引物,将正向引物和反向引物1:1混合后,95℃处理5~60min,再进行缓慢降温,形成sgrna的dna序列;
4)将crispr慢病毒表达载体进行双酶切,并与sgrna对应的双链dna进行连接,转入克隆感受态细胞,12h后,挑取单克隆进行测序,保留测序正确的克隆;
5)提取携带sgrna对应dna序列的crispr慢病毒载体质粒,进行病毒包装。
(十二)构建敲除原有tcr的crispr慢病毒载体
1)在pubmed上找到tcr基因的mrna的cds区,并分析tcr的保守区,将保守区进行敲除靶点的预测;
2)设计合成sgrna所需的正向引物和反向引物,将正向引物和反向引物1:1混合后,95℃处理5~60min,再进行缓慢降温,形成sgrna的dna序列;
3)将crispr慢病毒表达载体进行双酶切,并与sgrna对应的双链dna进行连接,转入克隆感受态细胞,12h后,挑取单克隆进行测序,保留测序正确的克隆;
4)提取携带sgrna对应dna序列的crispr慢病毒载体质粒,进行病毒包装。
(十三)tcr-t
1)复苏pbmc,以磁珠分选cd8+细胞,备用;
2)以步骤十一中获得的敲除免疫抑制信号分子的crispr慢病毒载体和步骤十二中获得的敲除原有tcr的crispr慢病毒载体,感染cd8+t细胞,同时进行免疫抑制性信号分子以及原有tcr的敲除;
3)感染后,cd8+t细胞在培养基中培养3天后,再转入步骤十构建的tcr基因表达载体,转入步骤实所得tcr基因;
4)以okm100+12%fbs将感染后的cd8+t细胞重悬,并置于刺激因子okm-25的预铺板上,记为第0天;
5)观察细胞情况和细胞密度,在第5天,将共培养细胞转移至大培养瓶中,补新鲜的培养液okm-100+12%fbs;
6)将细胞从75cm2瓶中那个转移至175cm2大瓶后培养液为okm-200+5%fbs;
7)培养至14-21天时,即可收获敲除免疫抑制性信号分子的tcr-t细胞,即rfft1细胞。
(十四)荷瘤小鼠生存实验
1)取工程细胞系接种nsg小鼠,做异位荷瘤动物模型。将5×105表达特异性抗原的肿瘤细胞悬于100μl生理盐水中,分别皮下注射至30只nsg小鼠的右侧胁肋部皮下,同时对小鼠进行编号。
2)在肿瘤生长至100-120mm3左右时分组回输细胞,根据肿瘤体积大小,将动物模型随机分成三组,每组5只小鼠,一组给予安慰剂生理盐水,一组给予没有进行任何遗传操作的t细胞(对照组)1×107,一组给予rfft1细胞1×107,首次注射细胞7天后进行第二次注射,7天之后第三次注射细胞,连续观察50天,统计存活数据,绘制生存曲线。
实验结果
1.突变位点及抗原表位预测
表1为测序检测到的突变位点及抗原表位预测结果,下划线标出的为突变的氨基酸。
表1抗原表位预测
2.抗原负载效率检测
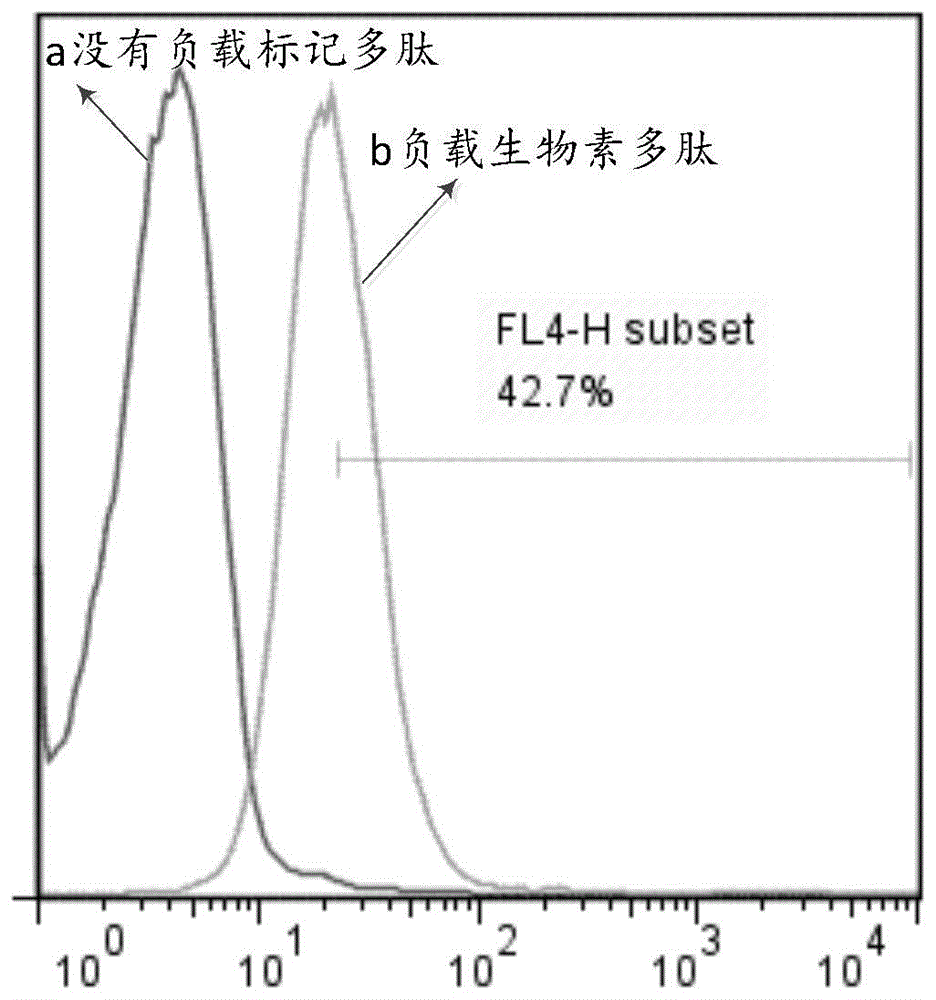
根据表1合成预测的突变抗原,并进行生物素的标记,抗原负载pbmc后,以pe标记的亲和链霉素检测生物素在细胞表面的分布情况,以检测pbmc提成多肽抗原的效率;结果如图1——抗原负载效率检测所示:a为没有负载标记多肽的检测结果,b为负载生物素多肽的检测结果,结果表明:pbmc的负载效率为42.7%(fl4-hsubset42.7%表示pe标记到的细胞占42.7%)。
3.筛选精准多肽
如图2所示,以12条多肽分别刺激培养的ff细胞,通过检测ifn-γ的分泌,检测有效的多肽,结果如图2所示:6号多肽引起的ifn-γ的释放量>高基线+系统误差,属于有效精准多肽。
4.rff细胞特异性细胞比例检测
以筛选的6号多肽,在ff细胞基础上进行多次刺激,培养后,以流式检测(facscaliburtm(bdbiosciences))对精准多肽特异性的t细胞比例,结果如图3所示,方框内为特异性t细胞:与无处理的对照组相比,经过多次冲击刺激后(rff细胞)可以释放ifn-γ的细胞比例明显高于没有多次冲击刺激的细胞(ff细胞)。说明,精准多肽多次刺激,可以提高特异性t细胞比例;同时以流式细胞仪进行cd8+ifn-γ+细胞(方框内)的分选。
5.高频tcr的鉴定和克隆
将分选得到细胞进行基因组的提取及tcr的测序,tcr的分布情况如图4所示(高频分布的前20),tcr16分布频率较高,说明,此tcr与突变抗原密切相关,根据tcr序列,对tcr进行扩增。
表2tcrβ链cdr3的序列情况
6.原有tcr的敲除
利用crispr技术,将pbmc上的原有的tcr进行敲除,流式检测敲除情况。结果如图5所示:可有效敲除原有tcr。
已知tcr-β:
氨基酸:
横线为cdr3序列,需要被替换的序列
替换后的tcr-β:
横线为替换的cdr3序列
7.免疫抑制性信号分子的敲除
利用crispr技术,将pbmc上的抑制性信号分子进行敲除,流式检测敲除信号分子敲除情况。结果如图6所示:可有阻断抑制性信号分子的表达。
8.特异性tcr表达情况
以流式检测tcr的表达情况,结果如图7所示:构建的tcr可以正常表达,转染3天,tcr+的细胞比例为48.3%;转染7天,tcr+的细胞比例为48.7%。
9.rfft1细胞对靶细胞的杀伤作用
分别以rff细胞、rfft细胞和rfft1细胞对突变抗原表位来源的靶细胞进行杀伤效率的检测,以无处理的细胞作为对照(mock),结果如图8所示,与对照组相比,rff细胞、rfft细胞和rfft1细胞对靶细胞均有一定杀伤效果,且在20:1(效应细胞:靶细胞)时,与mock组差异明显;整体趋势是,杀伤效率:rfft1细胞>rfft细胞>rff细胞;说明表达特异性tcr的t细胞,经过抑制性靶点的敲除可以有效的提高对肿瘤细胞的杀伤效率。
10.rfft1细胞释放细胞因子的检测
肿瘤细胞与效应细胞共培养时,由于效应细胞,可以识别肿瘤细胞上突变抗原,因此,会产生一系列的细胞因子,ifn-γ是抗肿瘤作用中最主要的细胞因子之一,图9为不同培养方案的细胞与肿瘤细胞共培养时,释放的ifn-γ的检测,结果表明:与效应细胞自身产生的ifn-γ相比(仅效应细胞),与肿瘤细胞共培养后,rff方案细胞、rfft方案细胞和rfft1细胞均可产生大量的ifn-γ,特别是rfft和rfft1细胞,由于表达了特异性tcr(rfft),同时抑制性信号已被敲除(rfft1),效应细胞可释放更多的ifn-γ,此结果与杀伤实验结果一致说明:表达特异性tcr的t细胞,经过抑制性靶点的敲除可以更有效的提高抗肿瘤能力。
11.荷瘤小鼠生存实验
结果如图10所示,回输本发明实施例的rfft1细胞对小鼠生存具有明显改善。p值小于0.01(p值=0.0008)表明具有统计学意义。
12.临床案例
给药过程:
第一疗程:每个月一次rfft1细胞,数量1×109个细胞,共2次;
第二疗程:每半年一次rfft1细胞,数量1×109个细胞,共2次。
表3
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!