一种具有抗炎作用的化合物及其制备方法和用途与流程
本发明属于药物合成,具体涉及一种具有抗炎作用的吡唑并[3,4-d]嘧啶类小分子衍生物及其制备方法和用途。
背景技术:
炎症与大多数疾病的发生和发展密切相关,包括冠心病、动脉粥样硬化、糖尿病和肿瘤等,炎症已经成为人类多种疾病的一个标志性特征。肿瘤坏死因子-α(tnf-α)、白细胞介素-6(il-6)、白细胞介素-1β(il-1β)等炎症因子在炎症发生发展过程中发挥着重要的作用。随着炎症信号传导途径研究的不断深入,il-6已经成为急慢性炎症有效的治疗靶点,在炎症性疾病治疗过程中扮演着重要角色。
目前,已开发出多种针对il-6信号级联的抑制剂,包括抗il-6、白介素-6受体(il-6r)和可溶糖蛋白130(sgp130fc)的单克隆抗体,其中部分抑制剂正在进行临床试验。使用单克隆抗体常伴随有再生障碍性贫血和充血性心力衰竭等不良反应。相比之下,小分子化合物药物与单克隆抗体药物相比较通常具有更好的生物利用度并且利于生产制造等特性。因此,开发新的il-6相关的小分子抑制剂具有非常好的前景。
吡唑并[3,4-d]嘧啶类化合物属于含氮稠环化合物,是嘌呤的的电子等排体,且由于它与嘌呤具有类似的结构从而显示出多种药理作用。因此,吡唑并[3,4-d]嘧啶类化合物一直是药物学领域的研究热点。已报道,吡唑并[3,4-d]嘧啶类化合物具有抗菌、抗病毒、抗癌、抗炎、抗高血压、抗糖尿病、抑制黄嘌呤氧化酶从而治疗痛风等多种药理活性。
但是,目前已知吡唑并[3,4-d]嘧啶类化合物在抗炎作用上还普遍存在抗炎效率低、毒副作用强的问题。所以,制备出更多结构新颖的il-6相关的小分子抑制剂具有非常重要的意义。
技术实现要素:
本发明的目的在于提供一种结构新颖的吡唑并[3,4-d]嘧啶类小分子衍生物。
本发明提供了一种式i所示的化合物、或其盐:
其中,r1为
r2选自卤素、氨基、羟基、羧基、被0~5个r4取代的以下基团:芳基、杂芳基、饱和环烷基、饱和杂环基、c1-6烷基、c1-6烷氧基;r4选自羟基、羧基、卤素、h、c1-6烷基、c1-6烷氧基;
r3选自被0~3个r6取代的以下基团:c1-8烷基、c1-8烷氧基、c2-8烯基、c2-8炔基;r6选自羟基、羧基、卤素。
进一步地,
r1为-wr5,其中,w选自nh或o,r5选自h、c1-4烷基、c1-4烷氧基、卤素、羟基、羧基;
r2为
r3选自c1-4烷基。
进一步地,
r1选自
r2选自
r3选自
进一步地,
所述化合物选自以下结构之一:
本发明还提供了一种制备上述化合物1-9的方法,所述方法包括以下步骤:
(1)化合物10与碘代异丙烷或碘代正丁烷反应,得到化合物11;
(2)化合物11与甲醇钠反应,得到化合物12;
(3)化合物12与3-羟基苯硼酸频哪醇酯或4-羟基苯硼酸频哪醇酯反应,即得化合物1、2、3或4;
或,所述方法包括以下步骤:
(2’)化合物11与甲胺反应,得到化合物13;
(3’)化合物13与3-羟基苯硼酸频哪醇酯或4-羟基苯硼酸频哪醇酯反应,即得化合物5或6;
或,所述方法包括以下步骤:
(a)化合物14与碘代异丙烷反应,得到化合物15;
(b)化合物15与3-羟基苯硼酸频哪醇酯或4-羟基苯硼酸频哪醇酯反应,即得化合物7或8;
或,所述方法包括以下步骤:
(a’)化合物16与叔丁肼盐酸盐反应,得到化合物17;
(b’)化合物17与甲酰胺反应,得到化合物18;
(c’)化合物18与n-溴代琥珀酰亚胺反应,得到化合物19;
(d’)化合物19与5-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)嘧啶-3-醇反应,即得化合物9;
上述化合物10的结构为
化合物13的结构为
化合物14的结构为
化合物16的结构为
进一步地,
步骤(1)中,所述反应是在碳酸钾的作用下进行的,反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(2)中,所述反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(2’)中,所述反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(3)或(3’)中,所述反应是在pdcl2dppf和碳酸钾的作用下进行的,反应溶剂为1,4-二氧六环与水的混合溶液,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(a)中,所述反应是在碳酸钾的作用下进行的,反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(b)中,所述反应是在pdcl2dppf和碳酸钾的作用下进行的,反应溶剂为1,4-二氧六环与水的混合溶液,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(a’)中,所述反应是在三乙胺的作用下进行的,反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(b’)中,所述反应温度为100~200℃,反应时间为2.0~5.0小时;
步骤(c’)中,所述反应溶剂为有机溶剂,反应温度为60~100℃,反应时间为2.0~5.0小时;
步骤(d’)中,所述反应是在pdcl2dppf和碳酸钾的作用下进行的,反应溶剂为1,4-二氧六环与水的混合溶液,反应温度为60~100℃,反应时间为2.0~5.0小时。
进一步地,
步骤(1)中,所述反应溶剂为mecn,反应温度为100℃,反应时间为3小时;
步骤(2)中,所述反应溶剂为甲醇,反应温度为65℃,反应时间为3.5小时;
步骤(2’)中,所述反应溶剂为乙醇,反应温度为75℃,反应时间为3.5小时;
步骤(3)或(3’)中,所述反应溶剂为体积比为4:1的1,4-二氧六环与水的混合溶液,反应温度为100℃,反应时间为2小时;
步骤(a)中,所述反应溶剂为mecn,反应温度为80℃,反应时间为3小时;
步骤(b)中,所述反应溶剂为体积比为4:1的1,4-二氧六环与水的混合溶液,反应温度为100℃,反应时间为2小时;
步骤(a’)中,所述反应溶剂为乙醇,反应温度为78℃,反应时间为3小时;
步骤(b’)中,所述反应温度为180℃,反应时间为5小时;
步骤(c’)中,所述反应溶剂为甲醇,反应温度为100℃,反应时间为2小时;
步骤(d’)中,所述反应溶剂为体积比为4:1的1,4-二氧六环与水的混合溶液,反应温度为100℃,反应时间为2小时。
本发明还提供了上述化合物、或其盐在制备白细胞介素-6抑制剂上的用途。
本发明还提供了上述化合物、或其盐在制备抗炎药物上的用途,优选地,所述抗炎药物能够降低磷酸化nf-κbp65蛋白水平、磷酸化akt蛋白水平、磷酸化stat3蛋白水平及粘附分子icam-1的蛋白水平。
本发明还提供了一种药物组合物,所述药物组合物是以上述化合物、或其盐为活性成分,加上药学上可接受的辅料制成。
实验结果表明,本发明的化合物,特别是化合物7和化合物9,能够有效抑制炎症因子il-6的释放水平;化合物9可抑制磷酸化nf-κbp65蛋白水平、磷酸化akt蛋白水平、磷酸化stat3蛋白水平及粘附分子icam-1的蛋白水平。本发明化合物在制备治疗炎症性疾病的药物上具有非常好的应用前景。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
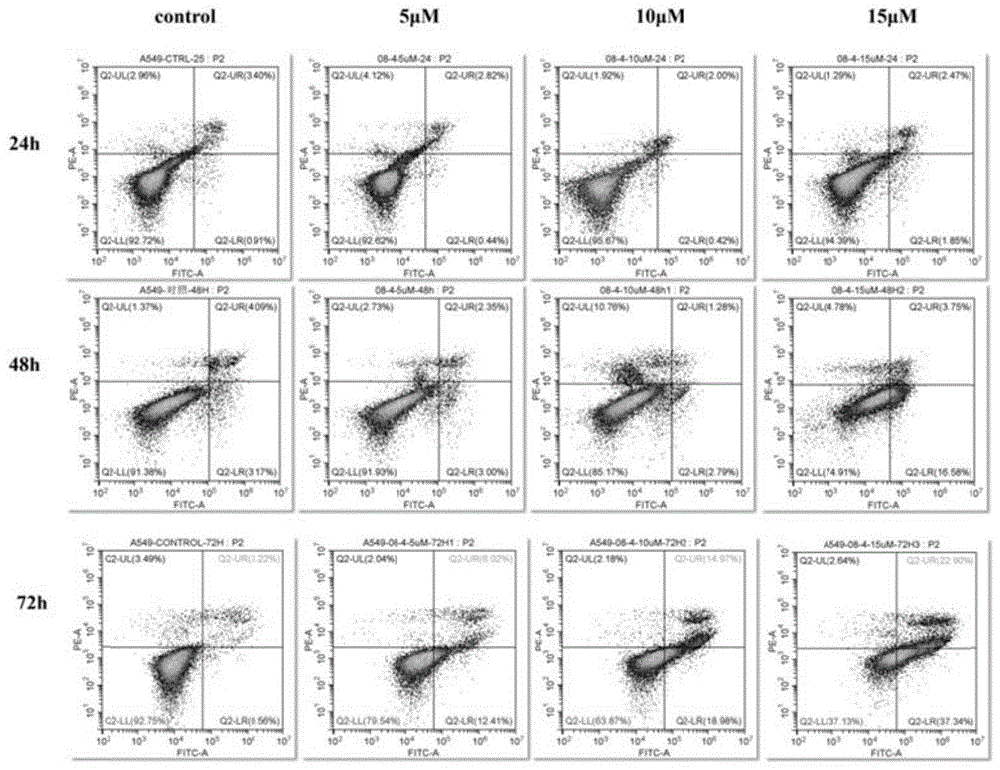
图1.化合物7的凋亡结果-流式图。
图2.化合物7对a549细胞周期的抑制-流式图。
图3.化合物9对a549细胞凋亡的影响-流式图。
图4.化合物9对a549细胞周期的抑制-流式图。
图5.化合物对炎症因子il-6水平的抑制作用(
图6.westernblotting法检测il-6上下游相关信号通路蛋白表达水平(
图7.westernblotting法检测icam-1蛋白表达水平,(
具体实施方式
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
其中,部分实验试剂来源如表1所示:
表1.部分实验试剂
主要仪器如下:
(1)薄层层析板:60f245型,0.2mm,merck,德国
(2)柱层析硅胶:硅胶h-gf254,200-300目,青岛海洋化工厂
(3)质谱分析仪:q-tof光谱仪,esi电离源,德国bruker
(4)核磁共振仪:avii-400mhz、avii-600mhz或avii-800mhz,tms为内标,德国bruker。
本发明采用下述路线,分别合成了本发明的目标化合物:
(1)吡唑并[3,4-d]嘧啶衍生物合成路线一:
试剂和条件:i)碘代异丙烷或碘代正丁烷,k2co3,mecn,100℃,3h,收率:11a:85%,11b:83%;ii)ch3ona,ch3oh,65℃,3.5h,收率:12a:73%,12b:80%;iii)甲胺,乙醇,75℃,3.5h,yield:69%;iv)频哪醇硼酸酯,pdcl2dppf,k2co3,1,4-二氧六环/水(4:1),100℃,2h,收率:~73%-85%。(2)吡唑并[3,4-d]嘧啶衍生物合成路线二:
试剂和条件:i)碘代异丙烷,k2co3,mecn,80℃,3h,收率:83%;ii)频哪醇硼酸酯,pdcl2dppf,k2co3,1,4-二氧六环/水(4:1),100℃,2h,yield:~46%-85%。
(3)吡唑并[3,4-d]嘧啶衍生物合成路线三:
试剂和条件:i)叔丁肼盐酸盐,三乙胺,乙醇,78℃,3h,收率:96%;ii)甲酰胺,180℃,5h,收率:91%;iii)n-溴代琥珀酰亚胺,mecn,100℃,2h,收率:89%;iv)频哪醇硼酸酯,pdcl2dppf,k2co3,1,4-二氧六环/水(4:1),100℃,2h,yield:47%。
本发明目标化合物的合成路径主要分成3种:①以3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶为起始原料;②以3-溴-4-氨基-1h-吡唑并[3,4-d]嘧啶为起始原料;③以2-乙氧基亚甲基丙二腈和叔丁肼盐酸盐为起始原料。当r3为取代异丙基或正丁基时,合成采用经典的烷基化取代反应,碘代异丙烷或碘代正丁烷在碳酸钾的催化下与吡啶并[3,4-d]嘧啶环发生亲核取代反应得到烷基取代的关键中间体。当r3为叔丁基取代时,由于叔丁基空间位阻较大,不能通过上述的合成路径与碘代叔丁烷反应得到产物。要想在r3位置引入叔丁基,需要在闭环前引入,参照todorovic等的合成方法,以吡唑环为母环合成吡唑并[3,4-d]嘧啶衍生物:首先2-乙氧基亚甲基丙二腈和叔丁肼盐酸盐反应生成1-叔丁基-5-氨基-1h-吡唑-4-腈,然后再与甲酰胺反应得到r3为叔丁基的吡唑并[3,4-d]嘧啶母核结构,前两步产率都较高(约90%),之后与n-溴代丁二酰亚胺(nbs)反应得到关键中间体,前三步综合产率约75%。在杂环上引入不同取代芳基r2时采用经典的suzuki偶联反应,采用碳酸钾及氯化钯作为催化剂与芳基取代频哪醇硼酸酯反应。
以下为本发明具体化合物的合成步骤:
实施例1、本发明化合物的制备
1-正丁基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶(11a):在250ml圆底烧瓶中加入3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶10(1g,4.3mmol)和150ml干燥乙腈溶液,搅拌溶解,依次加入碳酸钾(1.19g,8.6mmol)和碘代正丁基3ml(滴加),室温搅拌10分钟,然后将温度升至80℃回流。继续反应约3h,tlc监测表明反应完毕(展开剂:ch2cl2/ch3oh=95/5)。待反应液冷却后,加入50ml水和ch2cl2(100ml×3)萃取,合并有机相,加入适量无水硫酸钠干燥,过滤,浓缩。粗产物使用硅胶柱层析分离(洗脱剂:ch2cl2)得到白色固体产物1-正丁基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶1.06g,产率:85%。1hnmr(600mhz,cdcl3):δ0.93(t,j=7.4,3h),1.31(m,2h),1.90(m,2h),4.44(t,j=7.2,2h),8.71(s,1h).13cnmr(151mhz,cdcl3):δ13.69,19.97,31.61,48.11,112.36,119.27,153.87,155.22,155.31.
1-异丙基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶(11b):按照烷基化合成11a的方法:化合物103-溴-4-氯-1h-吡唑并[3,4-d]嘧啶(1g,4.3mmol)参与反应得到白色固体产物1-异丙基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶0.98g,产率:83%。1hnmr(600mhz,cdcl3):δ1.56(d,j=6.7,6h),5.18(hept,j=6.7,1h),8.70(s,1h).13cnmr(151mhz,cdcl3):δ22.11,50.80,112.55,119.04,153.06,154.93,155.19.
1-正丁基-3-溴-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(12a):将1-正丁基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶11a(0.5g,1.74mmol)溶于50ml干燥ch3oh溶液中,加入15ml2mch3ona溶液。该混合液在65℃约反应3.5h,tlc监测反应结束(展开剂:ch2cl2/ch3oh=95/5)。待反应液冷却至室温,加入30ml水和ch2cl2(150ml×3)萃取,合并有机相,加入适量无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离(洗脱剂:ch2cl2)得到白色固体产物1-正丁基-3-溴-4-甲氧基-1h-吡唑并[3,4-d]嘧啶0.36g,产率:73%。hrms-esi(m/z)calcdfor[m+h]+,285.0273;found,285.1153.
1-异丙基-3-溴-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(12b):采用合成化合物12a的方法,经反应,纯化得到12b,产率:80%。1hnmr(600mhz,cdcl3):δ1.53(d,j=6.7,6h),4.15(s,3h),5.11(hept,j=6.7,1h),8.48(s,1h).13cnmr(151mhz,cdcl3)δ22.19,50.16,54.62,103.19,117.72,154.56,155.69,164.03.
本发明中所涉及的在吡唑并[3,4-d]嘧啶环母核c9位置引入芳基这一过程,均采用经典的suzukicoupling反应。具体操作方法以合成化合物(1)为例。
1-正丁基-3-(3-酚基)-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(1):在装有75ml1,4-二氧六环/水(4:1)混合溶液的100ml圆底烧瓶中依次加入1-正丁基-3-溴-4-甲氧基-1h-吡唑并[3,4-d]嘧啶12a(0.20g,0.70mmol),3-羟基苯硼酸频哪醇酯(0.23g,1.05mmol),k2co3(0.19g,1.4mmol),pdcl2dppf(0.13g,0.18mmol),室温下充分搅拌至完全溶解。将反应置于油浴锅,加热至100℃回流,反应约2h,tlc监测反应结束(展开剂:ch2cl2/ch3oh=95/5),溶液由橙黄色变成黑色。待反应液冷却后,加入30ml水和ch2cl2(100ml×3)萃取,合并有机相,加入适量无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离(洗脱剂:ch2cl2/ch3oh=100/1-100/3)得到淡黄色固体产物1-正丁基-3-(3-酚基)-4-甲氧基-1h-吡唑并[3,4-d]嘧啶0.16g,产率:73.9%。1hnmr(600mhz,cdcl3):δ0.93(t,j=7.4,3h),1.36(m,2h),1.95(m,2h),4.16(s,3h),4.49(t,j=7.2,2h),6.91(dd,j=8.0,2.3,1h),7.33(t,j=7.9,1h),7.56(s,1h),7.63(d,j=7.7,1h),8.58(s,1h).13cnmr(151mhz,cdcl3):δ13.82,20.12,31.95,47.59,54.62,100.44,115.71,116.04,121.34,129.85,133.91,144.04,155.02,155.64,156.11,164.42.hrms-esi(m/z)calcdfor[m+h]+,299.1430;found,299.1505.
1-正丁基-3-(4-酚基)-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(2):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:85%。1hnmr(600mhz,dmso):δ0.88(t,j=7.4,3h),1.25(m,2h),1.84(m,3h),4.10(s,3h),4.40(t,j=6.9,2h),6.87(d,j=8.7,2h),7.84(d,j=8.6,2h),8.57(s,1h),9.71(s,1h).13cnmr(151mhz,dmso):δ13.86,19.76,31.51,46.73,54.75,99.35,115.66,123.39,130.13,143.71,155.34,155.75,158.45,164.00.hrms-esi(m/z)calcdfor[m+h]+,299.1430;found,299.1514.
1-异丙基-3-(3-酚基)-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(3):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:77%。1hnmr(600mhz,dmso):δ1.52(d,j=6.7,6h),4.11(s,3h),5.16(hept,j=6.8,1h),6.82(ddd,j=8.1,2.4,1.1,1h),7.28(t,j=7.7,1h),7.46(d,j=1.0,1h),7.47(s,1h),8.60(s,1h),9.57(s,1h).13cnmr(151mhz,dmso):δ21.78,48.80,54.37,99.40,115.00,115.60,119.13,129.38,133.29,142.80,154.49,154.73,157.31,163.44.hrms-esi(m/z)calcdfor[m+h]+,285.1273;found,285.1331.
1-异丙基-3-(4-酚基)-4-甲氧基-1h-吡唑并[3,4-d]嘧啶(4):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:83%。1hnmr(600mhz,dmso):δ1.51(d,j=6.7,6h),4.10(s,3h),5.13(m,1h),6.87(d,j=8.6,2h),7.83(d,j=8.6,2h),8.57(s,1h),9.72(s,1h).13cnmr(151mhz,dmso):δ21.77,48.64,54.28,99.15,115.17,123.08,129.69,143.03,154.41,154.66,157.94,163.49.hrms-esi(m/z)calcdfor[m+h]+,285.1273;found,285.1327.
1-异丙基-3-溴-4-甲氨基-1h-吡唑并[3,4-d]嘧啶(13):0.5g(1.83mmol)1-异丙基-3-溴-4-氯-1h-吡唑并[3,4-d]嘧啶11b溶于100ml无水乙醇溶液,室温搅拌下加入40%甲胺水溶液0.8ml,然后加热至回流,约3.5h,tlc监测反应结束(展开剂:pe/etoac=4:1.5)。待反应液冷却至室温,30ml水和etoac(100ml×3)萃取,合并有机相,加入适量无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离纯化(洗脱剂:pe/etoac=100/5-100/10)得到白色固体产物1-异丙基-3-溴-4-甲氨基-1h-吡唑并[3,4-d]嘧啶0.34g,产率:69%。hrms-esi(m/z)calcdfor[m+h]+,270.0276;found,270.0354.
1-异丙基-3-(3-酚基)-4-甲氨基-1h-吡唑并[3,4-d]嘧啶(5):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:75%。1hnmr(600mhz,dmso):δ1.45(d,j=6.8,6h),2.93(d,j=4.7,3h),4.96(hept,j=6.6,1h),5.53(d,j=4.8,1h),6.74(dd,j=8.1,1.6,1h),6.86(d,j=8.5,1h),7.25(t,j=7.8,1h),7.39(s,1h),8.22(s,1h),9.55(s,1h).13cnmr(151mhz,dmso):δ22.38,27.79,45.29,100.11,113.66,115.15,118.98,119.83,130.03,136.26,148.81,151.13,156.69,157.80.hrms-esi(m/z)calcdfor[m+h]+,284.1433;found,284.1511.
1-异丙基-3-(4-酚基)-4-甲氨基-1h-吡唑并[3,4-d]嘧啶(6):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:84%。1hnmr(600mhz,dmso):δ1.46(d,j=6.7,6h),2.94(d,j=4.7,3h),5.03(m,1h),6.25(dd,j=9.1,4.5,1h),6.91(d,j=8.5,2h),7.45(d,j=8.5,2h),8.29(s,1h),9.74(s,1h).13cnmr(151mhz,dmso):δ21.80,27.93,47.92,97.63,115.91,123.95,129.58,143.08,152.64,155.14,157.20,157.81.hrms-esi(m/z)calcdfor[m+h]+,284.1433;found,284.1506.
1-异丙基-3-溴-4-氨基-1h-吡唑并[3,4-d]嘧啶(15):按照烷基化合成11a的方法得到15,产率:83%。hrms-esi(m/z)calcdfor[m+h]+,256.0120;found,256.0197.
1-异丙基-3-(3-酚基)-4-氨基-1h-吡唑并[3,4-d]嘧啶(7):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:76%。1hnmr(600mhz,dmso):δ1.48(d,j=6.7,6h),5.05(m,1h),6.87(dd,j=8.2,1.4,1h),7.07(s,1h),7.08(d,j=2.9,1h),7.34(t,j=8.0,1h),8.23(s,1h),9.70(s,1h).13cnmr(151mhz,dmso):δ21.78,48.04,97.35,114.92,115.69,118.85,130.28,134.38,143.26,153.21,155.41,157.83,158.01.hrms-esi(m/z)calcdfor[m+h]+,270.1277;found,270.1357.
1-异丙基-3-(4-酚基)-4-氨基-1h-吡唑并[3,4-d]嘧啶(8):通过suzukicoupling反应,具体操作见化合物1合成方法,产率:85%。1hnmr(600mhz,dmso):δ1.47(d,j=6.7,6h),5.03(hept,j=6.7,1h),6.92(d,j=8.5,2h),7.47(d,j=8.5,2h),8.21(s,1h),9.76(s,1h).13cnmr(151mhz,dmso):δ21.79,47.92,97.33,115.89,123.85,129.59,143.46,153.13,155.27,157.88,158.08.hrms-esi(m/z)calcdfor[m+h]+,270.1277;found,270.1360.
1-叔丁基-5-氨基-1h-吡唑-4-腈(17):将叔丁肼盐酸盐(8g,64.20mmol)和三乙胺(6.50g,64.20mmol)溶于500ml无水乙醇中,然后在搅拌下向反应瓶中慢慢滴加2-乙氧基亚甲基丙二腈(7.83g,64.20mmol)。加热至78℃回流,约3h后,tlc监测反应结束(展开剂:pe/etoac=1:1)。待反应液冷却至室温,减压浓缩除去溶剂,加入100ml水和ch2cl2(300ml×3)萃取,合并有机相,加入适量无水硫酸钠干燥,浓缩得到橙黄色粘稠固体。将得到的粗产物溶于60ml乙酸乙酯-正己烷(1:9)混合溶液,超声约5min,静止,观察有大量晶形沉淀产生,过滤(乙酸乙酯-正己烷混合溶液洗3遍),烘干得到橙色固体产物1-叔丁基-5-氨基-1h-吡唑-4-腈10.11g,产率:96%。1hnmr(600mhz,dmso):δ1.50(s,9h),6.22(s,2h),7.44(s,1h).13cnmr(151mhz,dmso):δ28.22,58.75,74.54,115.23,138.00,150.71.hrms-esi(m/z)calcdfor[m+na]+,187.1062;found,187.0968.
1-叔丁基-4-氨基-1h-吡唑并[3,4-d]嘧啶(18):在100ml圆底烧瓶中加入1-叔丁基-5-氨基-1h-吡唑-4-腈(5g,30.47mmol),甲酰胺溶液80ml,混合液置于油浴锅加热至180℃,约5h后,tlc监测反应结束(展开剂:pe/etoac=1:1)。待反应液冷却后,加入50ml水和ch2cl2(200ml×3)萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离纯化(洗脱剂:pe/etoac=4/1-1/1)得到白色固体产物1-叔丁基-4-氨基-1h-吡唑并[3,4-d]嘧啶5.82g,产率:91%。1hnmr(600mhz,dmso):δ=1.69(s,9h),8.03(s,1h),8.14(s,1h).13cnmr(151mhz,dmso):δ28.76,59.22,101.42,130.03,152.71,154.71,158.16.hrms-esi(m/z)calcdfor[m+h]+,192.1171;found,192.1254.
1-叔丁基-3-溴-4-氨基-1h-吡唑并[3,4-d]嘧啶(19):称取1-叔丁基-4-氨基-1h-吡唑并[3,4-d]嘧啶183g(15.70mmol)溶于250ml无水乙腈,室温搅拌下加入n-溴代琥珀酰亚胺(nbs,4.19g,23.55mmol)直至完全溶解。加热至100℃回流约2h,tlc监测反应结束(展开剂:ch2cl2/ch3oh=95/5),待反应液冷却至室温,二氯甲烷/水萃取(×3),合并有机相,适量无水硫酸钠干燥,过滤,浓缩,硅胶柱层析分离纯化(洗脱剂:ch2cl2)得到白色固体产物1-叔丁基-3-溴-4-氨基-1h-吡唑并[3,4-d]嘧啶4.22g,产率:89%。1hnmr(600mhz,dmso):δ1.67(s,9h),8.20(s,1h).13cnmr(151mhz,dmso):δ28.60,60.60,100.45,115.28,153.51,155.63,157.50.hrms-esi(m/z)calcdfor[m+h]+,270.0276;found,270.0355.
1-叔丁基-3-(5-羟基-3-吡啶基)-4-氨基-1h-吡唑并[3,4-d]嘧啶(9):通过suzukicoupling反应,具体操作详见化合物1合成方法,1-叔丁基-3-溴-4-氨基-1h-吡唑并[3,4-d]嘧啶(1.45g,5.43mmol)参与反应得到灰色固体产物1-叔丁基-3-(5-羟基-3-吡啶基)-4-氨基-1h-吡唑并[3,4-d]嘧啶0.58g,产率:47%。其中展开剂:ch2cl2/ch3oh=10/1,洗脱剂:ch2cl2/ch3oh=100:1-100:5。1hnmr(600mhz,dmso):δ1.75(s,9h),7.38(dd,j=2.5,2.0,1h),8.20(d,j=2.7,1h),8.25(s,1h),8.29(d,j=1.8,1h),10.16(s,1h).13cnmr(151mhz,dmso)δ29.16,60.32,99.29,121.91,130.11,138.39,139.21,140.03,154.11,154.47,155.17,158.73.hrms-esi(m/z)calcdfor[m+h]+,285.1386;found,285.1457.
通过高分辨质谱、核磁共振氢谱、核磁共振碳谱分析手段,得到前述鉴定数据,通过分析数据,确认制得的上述化合物1-9的结构如下:
实验结果证明,本发明制备得到了化合物1-9。
以下通过实验例证明本发明化合物的有益效果:
炎症因子il-6是急慢性炎症有效的治疗靶点,在炎症性疾病治疗过程中扮演着重要角色。本发明参照müller等人的方法,以尿苷作为阳性对照,采用il-4诱导人肺癌细胞a549释放il-6细胞炎症模型评价合成化合物的il-6下调水平。并对化合物的细胞增殖抑制活性、细胞凋亡与周期等生物活性进行了评价。
一、实验方法
1实验材料及仪器:
1.1实验所需部分材料及试剂列于表2。
表2.部分实验材料与试剂
1.2生物实验主要仪器:
(1)co2培养箱:thermo311型,thermo,美国
(2)荧光倒置显微镜:te2000-u型,nikon,日本
(3)流式细胞仪:facsvalibur,bd公司,美国
(4)全自动扫描式多功能酶标仪:multiskansky,thermo,美国
(5)4℃/-20℃冰箱:hxc-158,海尔公司,中国
(6)-80℃超低温冰箱:forma,thermo,美国
(7)琼脂糖凝胶电泳仪:e-gelpowersnap,invitrogen,美国
(8)超声波细胞破碎仪:jyd-650,上海之信仪器有限公司,中国
(9)恒温摇床:sph-211b-gz,上海世平实验有限公司,中国
(10)全自动凝胶成像系统:geldoc,bio-rad公司,美国
(11)台式微量冷冻离心机:22r,beckman,美国
(12)高速低温离心机:d37520,heraeus,德国
(13)细胞活力分析仪:vi-cell,beckman,美国
(14)循环恒温水浴箱:zsxh-618/625,上海智城分析仪器制造有限公司
1.3超分子结构及性质研究表征仪器:
(1)x射线单晶衍射分析:德国brukerapex-iiccd衍射仪
(2)热分析(dsc/tga):荷兰mettlertoledotga/dsc1同步热分析仪
(3)变温多晶粉末x射线衍射分析(vt-pxrd):荷兰panalytical-empyrean型x-射线衍射仪
(4)扫描电镜(sem):高分辨inspectf50型扫描电子显微镜
(5)紫外-可见吸收光谱分析:uv-3600分光光度计
2细胞培养
将具有类人肺泡上皮化a549细胞培养在rpmi1640培养基,并补充加入10%胎牛血清(fbs)和1%青链霉素。分装到培养瓶中,拧紧瓶盖,适度拧紧后稍回转,以利于co2的进入,将细胞孵育于37℃,5%co2培养箱。其中a549细胞购买于中科院细胞库(中国)。
3细胞增殖抑制实验(mtt)
用0.25%胰蛋白酶将贴壁细胞消化下来,用含10%胎牛血清的rpmi1604培养液配成单个细胞悬液,以每孔2000个细胞的密度接种于96孔板中,每孔体积100ul。将培养板移入co2孵箱中,在37℃、5%co2条件下,孵育24小时。将不同浓度的药物加入每孔中,设置3个复孔,然后继续培养72小时。按操作手册每孔加入溴化噻唑蓝四氮唑(mtt)20μl,继续孵育4小时,终止培养,小心吸弃孔内培养上清液。选择490nm波长,在酶联免疫检测仪上调定各孔收值,记录结果。以时间为横轴,光吸收值为纵轴绘图。并用bliss法计算半数抑制浓度(ic50)。每个实验至少重复3次。
4细胞周期检测(flowcytometry)
细胞周期测定采用流式检测方法,首先pi(碘化丙啶)标记染色,然后通过流式细胞仪检测pi从而测定细胞群体的周期分布。a549细胞加入含edta的胰酶进行消化,待消化后,加入含10%fbs的dmem终止消化,轻轻吹打细胞,将细胞悬液收集到离心管中。1000rpm/min,离心10分钟,弃上清。逐滴加入5ml预冷的70-80%的乙醇,涡旋以混匀细胞,4℃避光过夜。1000-1500rpm/min,离心10分钟,弃上清。然后依次用pbs、含pi的pbs溶液清洗细胞2次以去除所有的乙醇。将细胞重悬于0.5mlpi/rnase染色液,室温避光孵育30分钟。轻轻吹打混匀,过滤至流式管中,4℃避光保存样本,1小时之内上流式细胞仪检测。
5细胞凋亡检测(flowcytometry)
6孔板培养a549细胞,分别加入不同浓度的化合物和dmso作为空白对照。24小时后,使用不含edta的0.25%胰蛋白酶收集细胞,pbs溶液冲洗2次。然后使用细胞凋亡检测试剂盒按照说明书进行染色。之后用流式细胞仪检测分析。
6酶联免疫吸附实验(elisa)
采用双抗体夹心(sandwich)法检测样品中靶蛋白的浓度。分别将样品或不同浓度标准品按照100μl/孔加入相应孔中,用封板膜(透明)封住反应孔,室温孵育120分钟。pbst洗板5次,且最后一次置于厚吸水纸上拍干。加入生物素化抗体100μl/孔,用封板膜封住反应孔,室温孵育60分钟。洗板5次,同样最后一次厚吸水纸上拍干。加入辣根过氧化物酶标记streptavidin(hrp-streptavidin)100μl/孔,封板膜(白色)封住反应孔,室温避光孵育20分钟。洗板5次,最后一次拍干。加入显色剂tmb溶液100μl/孔,封板膜(白色)封住反应孔,室温避光孵育20分钟,至标准品和样品出现非常显著的颜色变化。加入终止液50μl/孔,混匀后立即测量450nm处吸光度值。il-6采用特定的elisa试剂盒按照说明书进行测试。
7蛋白免疫印迹分析(westernblotting)
1)sds--page凝胶的制备
sds--page凝胶的制备以配制10%tris-甘氨酸sds-page为例:按照双重蒸馏水3.18ml,30%聚丙烯酰胺溶液2.66ml,1.5mtris-hcl溶液(ph8.8)2ml,10%过硫酸铵0.08ml,10%十二烷基硫酸钠(sds)溶液0.08ml,四甲基二乙胺(temed)溶液(临用前加入)0.0032ml的方法制备。
2)sds--page电泳
将制备好的凝胶板固定于电泳装置上,并将其下方的挡板打开。加入tris--甘氨酸电泳缓冲工作液,排除凝胶底部的气泡,按照预定的顺序加样,然后与电源连接。电源设置:100v,400a,约2小时后当溴酚蓝到达分离胶底部时,关闭电源并取出玻璃板停止电泳。
3)转膜
依次放入多孔垫片浸湿后的转膜夹子、精确滤纸两张、分离胶、pvdf膜、精确滤纸两张;膜、滤纸与凝胶之间需对齐且不能有气泡。将夹子放入转膜电泳槽中,在电泳槽中加入4℃的caps缓冲液,打开冷凝自来水及搅拌器,连接电源进行转膜。电源设置:50v,400ma。约2.5小时后转膜结束。
4)免疫染色
将转膜后的pvdf膜稍浸泡于纯甲醇溶液,再放入tbs缓冲液中冲洗。加入封闭缓冲液(tbst)置于摇床室温封闭2小时。tbst溶液洗膜(4×10min),加入1:300或1:500稀释的一抗(溶于封闭缓冲液中),然后放在摇床上室温孵育2小时。tbst溶液洗膜(3×5min),加入1:1000稀释的二抗(溶于封闭缓冲液中)置于平缓摇动的摇床室温孵育1.5小时后用tbst溶液洗膜(3×5min),ecl化学发光试剂盒染色1分钟进行x光胶片曝光或扫图。
8统计分析
所有实验结果采用spss19.0软件进行统计学分析,以
二、实验结果
1、mtt法测定本发明化合物对a549细胞增殖抑制的影响
采用mtt法,分别测试不同浓度的化合物作用于人肺癌细胞a549细胞24h后细胞的抑制率,化合物浓度分别为3.125μm,6.25μm,12.5μm,25μm,50μm。化合物ic50值列于下表(表3)。可以看出,化合物7、化合物8、化合物9表现出一定的细胞增殖抑制活性,药物作用24h后测定ic50值分别为41.01μm,44.81μm,69.62μm。
表3.化合物作用于a549细胞的ic50值
2、细胞凋亡及细胞周期测定(flowcytometry)
对化合物7和化合物9进行了细胞周期测定并检测了细胞凋亡情况(图1-4,表4-5)。实验选用人肺癌细胞a549细胞作为检测系统,对照组为空白对照(dmso),化合物测试浓度分别为5μm、10μm、15μm,化合物作用于人肺癌细胞a549细胞24h、48h、72h后采用流式细胞术进行检测。在细胞凋亡结果图中,象限q1中的细胞为坏死细胞,象限q2为晚期凋亡细胞,象限q3区域中的细胞为早期凋亡细胞,象限q4中的细胞代表活细胞。
从测试结果可以看出,化合物9对细胞的影响较小,当化合物浓度为15μm,72h引起细胞总凋亡也只有15.66%。
另外,当化合物作用于a549细胞72h后,与对照组相比,化合物7具有较为明显的诱导a549细胞凋亡的作用并呈现出剂量依赖性(图1)。提示化合物7对a549细胞的抗增殖活性可能与其诱导细胞凋亡的能力有关,表明化合物7可能具有一定的抗癌活性。
表4.化合物7对a549细胞凋亡的影响
表5.化合物9对a549细胞凋亡的影响
3、elisa法测定化合物对il-4诱导a549细胞分泌il-6水平的影响
化合物作用下炎症因子il-6水平测定结果见图5,化合物浓度分别为1μm,2.5μm,5μm,10μm。
实验结果显示,不同浓度的化合物7、9作用于细胞炎症模型24h后,il-6水平均有一定程度降低,其中化合物9的抑制效果最强。化合物7和9均显示出良好的剂量依赖性,随着浓度的增加其抗炎活性逐渐增强。在浓度达到5μm后,化合物下调il-6的效能强于阳性对照药尿苷(uridine),开始表现较好的抗炎效果。
结合化合物的细胞凋亡及周期实验结果可知,本发明化合物对il-6释放水平的抑制不受细胞凋亡的影响。
4、炎症相关信号通路探究(westernblotting)
分别测试化合物作用后的磷酸化nf-κbp65蛋白水平,磷酸化stat3(ser727)蛋白水平和磷酸化akt(ser473)蛋白水平,采用westernblotting法检测这些通路中关键调节因子的变化。其中β-actin作为内参。本发明还测了化合物作用后粘附分子icam-1的蛋白表达。
结果显示,化合物9作用后与对照组相比,药物组磷酸化nf-κbp65蛋白水平、磷酸化stat3(ser727)蛋白水平及磷酸化akt(ser473)蛋白水平降低(图6)。粘附分子icam-1的蛋白水平也呈剂量依赖性降低(图7)。因此,本发明化合物9显示出的显著的抗炎活性,作为一种潜在的治疗炎症性疾病的药物具有非常好的应用前景。
综上,本发明制备得到了一种吡唑并[3,4-d]嘧啶类小分子衍生物,实验发现,本发明的化合物7和化合物9,特别是化合物9,能够有效抑制炎症因子il-6的释放水平,化合物9可抑制磷酸化nf-κbp65蛋白水平、磷酸化akt蛋白水平、磷酸化stat3蛋白水平及粘附分子icam-1的蛋白水平。因此,本发明化合物在制备治疗炎症性疾病的药物上具有非常好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!