一种含氮杂环取代的吲哚硫醚类化合物的制备方法与流程
本发明属于有机合成化学技术领域,具体涉及一种含氮杂环取代的吲哚硫醚类化合物的制备方法。
背景技术:
含氮杂环取代的吲哚硫醚类化合物是一类重要的中间体,其在医药、农药、化工等领域有着广泛的应用。目前,含氮杂环取代的吲哚硫醚类化合物的合成方法主要以过渡金属催化反应构建c-s键进行合成,如中国专利cn107698558和文献(rscadv,2015,5,39358-39365)报道了铜催化的合成方法;文献(jorgchem,2015,80(1):367-374)报道了钯催化合成方法。但此类方法所用的原料和催化剂来源少、价格昂贵,需要在高温下反应,反应条件苛刻,并且会造成过渡金属在产品中残留,不利于大量制备。因此,发展一种经济实用性高、反应条件温和、原料易得,且操作简便的方法用于合成含氮杂环取代的吲哚硫醚类化合物具有极其重要的意义。
技术实现要素:
本发明的目的在于,克服现有技术中存在的缺陷,提供一种含氮杂环取代的吲哚硫醚类化合物的制备方法,该方法不需要使用过渡金属催化,具有操作步骤简便、绿色环保、对底物的兼容性好、收率高等特点,适于含氮杂环取代的吲哚硫醚类化合物的工业化生产。
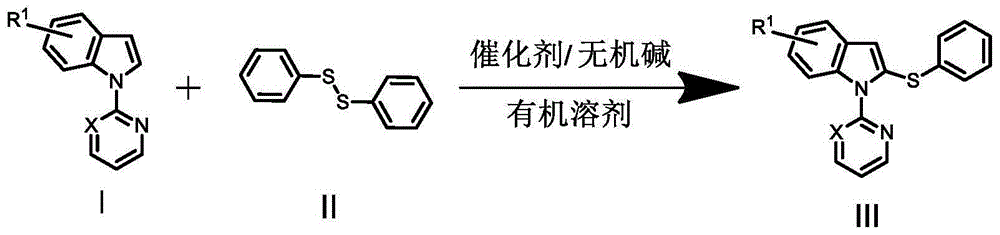
为实现上述目的,本发明的技术方案是设计一种含氮杂环取代的吲哚硫醚类化合物的制备方法,在一定的反应温度下,以式ⅰ所示的含氮杂环取代吲哚衍生物和式ⅱ所示的硫化试剂为原料,在催化剂和无机碱的存在下于有机溶剂中充分反应,得到所述的含氮杂环取代的吲哚硫醚类化合物,其结构通式如式ⅲ所示,其反应式为:
式ⅰ中,x选自ch或n中的一种;式ⅰ或ⅲ中,r1选自h或c1~c5烷基中的一种。
优选的技术方案是,所述的催化剂为碘单质。
优选的技术方案还有,所述的硫化试剂为二苯二硫醚。
优选的技术方案还有,所述的有机溶剂选自二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、1,2-二氯乙烷、四氢呋喃或乙腈中的一种。
优选的技术方案还有,所述的无机碱选自磷酸钾、碳酸钾、碳酸钠、碳酸铯、碳酸氢钾或碳酸氢钠中的一种。
优选的技术方案还有,所述的含氮杂环取代吲哚衍生物和硫化试剂的摩尔比为1:1~1:1.5。
进一步优选的技术方案还有,所述的含氮杂环取代吲哚衍生物和硫化试剂的摩尔比为1:1.1。
优选的技术方案还有,所述的反应温度为60~100℃。
进一步优选的技术方案还有,所述的反应温度为80℃。
本发明的优点和有益效果在于:
本发明方法以含氮杂环取代吲哚衍生物、硫化试剂为反应原料,在碘单质和无机碱存在的有机溶剂中催化合成含氮杂环取代的吲哚硫醚类化合物,该方法不需要使用过渡金属催化,具有操作步骤简便、绿色环保、对底物的兼容性好、收率高等特点,适于含氮杂环取代的吲哚硫醚类化合物的工业化生产。
附图说明
图1是本发明所述的含氮杂环取代的吲哚硫醚类化合物的合成路线;
图2是实施例1中制备的3-甲基-2-(苯硫基)-1-(吡啶-2-基)-1h-吲哚的核磁图谱;
图3是实施例4中制备的7-甲基-2-(苯硫基)-1-(嘧啶-2-基)-1h-吲哚的核磁图谱;
图4是实施例5中制备的2-(苯硫基)-1-(吡啶-2-基)-1h-吲哚的核磁图谱;
图5是实施例6中制备的3-甲基-2-(苯硫基)-1-(嘧啶-2-基)-1h-吲哚的核磁图谱。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
实施例1
采用本发明方法合成3-甲基-2-(苯硫基)-n-(2-吡啶基)-1h-吲哚,其反应式为:
具体合成步骤为:将3-甲基-n-(2-吡啶基)-1h-吲哚(1.04g,5mmol)和二苯基二硫醚(1.2g,5.5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、磷酸钾(2.1g,10mmol)及15ml二甲基亚砜,加热至80℃反应6小时,薄层色谱监测反应完全,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.42g(目标产物),收率90.2%(以3-甲基-n-(2-吡啶基)-1h-吲哚计),目标产物的核磁图谱参见图2,核磁数据如下:
1hnmr(300mhz,dmso-d6)δ8.58(s,1h),7.94~7.97(m,1h),7.71(d,j=6.0hz,1h),7.52(d,j=3.0hz,1h),7.39~7.44(m,4h),7.26~7.31(m,2h),7.18~7.22(m,3h),7.10(d,j=3.0hz,1h),6.91(d,j=3.0hz,2h),2.39(s,3h)。
实施例2
采用本发明方法合成3-甲基-2-(苯硫基)-n-(2-吡啶基)-1h-吲哚,具体合成步骤为:将3-甲基-n-(2-吡啶基)-1h-吲哚(1.04g,5mmol)和二苯基二硫醚(1.1g,5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、碳酸钾(1.4g,10mmol)及15mln,n-二甲基甲酰胺,加热至100℃反应6小时,薄层色谱监测反应完全,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.18g(目标产物),收率75.3%(以3-甲基-n-(2-吡啶基)-1h-啶基)-1h-吲哚计)。
实施例3
采用本发明方法合成3-甲基-2-(苯硫基)-n-(2-吡啶基)-1h-吲哚,具体合成步骤为:将3-甲基-n-(2-吡啶基)-1h-吲哚(1.04g,5mmol)和二苯基二硫醚(1.65g,7.5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、碳酸氢钾(1.5g,15mmol)及15ml1,2-二氯乙烷,加热至60℃反应12小时,薄层色谱监测反应完全,浓缩,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.03g(目标产物),收率65.2%(以3-甲基-n-(2-吡啶基)-1h-啶基)-1h-吲哚计)。
实施例4
采用本发明方法合成7-甲基-2-(苯硫基)-n-(2-嘧啶基)-1h-吲哚,其反应式为:
具体合成步骤为:将7-甲基-n-(2-嘧啶基)-1h-吲哚(1.05g,5mmol)和二苯基二硫醚(1.2g,5.5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、磷酸钾(2.1g,10mmol)及15ml二甲基亚砜,加热至80℃反应6小时,薄层色谱监测反应完全,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.35g(目标产物),收率85.8%(以7-甲基-n-(2-嘧啶基)-1h-吲哚计),目标产物的核磁图谱参见图3,核磁数据如下:
1hnmr(300mhz,dmso-d6)δ8.93(s,1h),7.64(t,j=3.0hz,1h),7.53(d,j=6.0hz,1h),7.22~7.24(m,2h),7.16~7.19(m,1h),7.09~7.11(m,3h),7.02~7.03(m,2h),1.78(s,3h)。
实施例5
采用本发明方法合成2-(苯硫基)-n-(2-吡啶基)-1h-吲哚,其反应式为:
具体合成步骤为:将n-(2-吡啶基)-1h-吲哚(0.97g,5mmol)和二苯基二硫醚(1.2g,5.5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、磷酸钾(2.1g,10mmol)及15ml二甲基亚砜,加热至80℃反应6小时,薄层色谱监测反应完全,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.4g(目标产物),收率92.4%(以n-(2-吡啶基)-1h-吲哚计),目标产物的核磁图谱参见图4,核磁数据如下:
1hnmr(300mhz,dmso-d6)δ8.59(s,1h),7.98~8.00(m,1h),7.67(d,j=6.0hz,1h),7.51(d,j=3.0hz,1h),7.44~7.47(m,1h),7.35(d,j=3.0hz,1h),7.22~7.24(m,3h),7.15~7.19(m,2h),7.06(d,j=3.0hz,1h),7.04(s,1h)。
实施例6
采用本发明方法合成3-甲基-2-(苯硫基)-n-(2-嘧啶基)-1h-吲哚,其反应式为:
具体合成步骤为:将3-甲基-n-(2-嘧啶基)-1h-吲哚(1.05g,5mmol)和二苯基二硫醚(1.2g,5.5mmol)置于反应瓶中,加入碘(0.13g,0.5mmol)、磷酸钾(2.1g,10mmol)及15ml二甲基亚砜,加热至80℃反应6小时,薄层色谱监测反应完全,加水30ml和乙酸乙酯20ml萃取,其中有机层依次经饱和食盐水洗涤、无水硫酸钠干燥、浓缩得到油状物,柱层析纯化得到无色油状物1.41g(目标产物),收率89.2%(以3-甲基-n-(2-嘧啶基)-1h-吲哚计),目标产物的核磁图谱参见图5,核磁数据如下:
1hnmr(300mhz,dmso-d6)δ8.92(s,1h),7.95(d,j=3.0hz,1h),7.69(d,j=6.0hz,1h),7.46~7.48(m,1h),7.38~7.41(m,1h),7.33~7.36(m,1h),7.25~7.28(m,1h),7.20~7.23(m,2h),7.09~7.12(m,1h),6.97(d,j=3.0hz,1h)。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!