柠檬醛及其衍生物在制备MRSA感染性疾病药物中的新用途的制作方法
本发明涉及柠檬醛及其衍生物的新用途,具体地,是在制备治疗葡萄球菌感染性疾病的药物中的用途。
背景技术:
耐甲氧西林金葡菌(methicillin-resistant Staphylococcus aureus,MRSA)被称作“超级细菌”,自1961年在英国被首次发现以来,即以惊人的速度在世界范围内蔓延,目前MRSA感染已超过艾滋病、结核和病毒性肝炎成为患者首位致死病因,严重威胁公共卫生安全。虽然利奈唑胺、达托霉素、头孢洛林、奥利万星、达巴万星、磷酸泰地唑胺等新药陆续获得FDA批准用于MRSA的治疗,但近年来临床上已发现上述药物的耐药菌株,且耐药率呈逐年上升趋势,故新型抗MRSA感染药物的研发显得尤为迫切。
柠檬醛(3,7-二甲基-2,6-辛二烯-1-醛)是一种单萜化合物,分子式为C10H16O,无色或淡黄色透明液体。柠檬醛通常由互为异构体的香叶醛(反式柠檬醛,柠檬醛a)和橙花醛(顺式柠檬醛,柠檬醛b)组成。天然柠檬醛主要存在于山苍子油、柠檬草油、马鞭草油、垂叶香茅油、草果油等植物精油中。
柠檬醛具有杀虫、驱避、抗菌、抗炎等广泛的药理活性。余伯良等,“柠檬醛对花生酱的防腐效果”中国调味品2002第3期公开了柠檬醛具有抗霉菌的作用。吕昭萍等,“山苍子油的抗菌作用研究”皮肤病与性病1997年第19期公开了含有80-90%柠檬醛的山苍子油具有广谱抗菌的作用。
技术实现要素:
本发明的技术方案提供了柠檬醛或其衍生物的用途。
本发明提供了柠檬醛或其衍生物在制备治疗耐药葡萄球菌感染性疾病的药物中的用途。
其中,所述耐药葡萄球菌为耐甲氧西林葡萄球菌。
其中,所述耐甲氧西林葡萄球菌为耐甲氧西林金黄色葡萄球菌;优选地,所述耐甲氧西林金黄色葡萄球菌为耐甲氧西林金黄色葡萄球菌ATCC43300或耐甲氧西林金黄色葡萄球菌ATCC33591。
其中,所述的药物是抑制耐药葡萄球菌生长或杀灭耐药葡萄球菌的药物。
本发明提供了一种治疗耐药葡萄球菌感染性疾病的药物,它是由柠檬醛或其衍生物为活性成分,加上药学上可接受的辅料或辅助性成分制备而成的制剂。
其中,所述制剂为注射制剂或口服制剂。
本发明提供了一种治疗耐药葡萄球菌感染性疾病的联合用药物,它含有相同或不同规格单位制剂的用于同时或者分别给药的柠檬醛和治疗耐药葡萄球菌感染性疾病的药物,以及药学上可接受的载体。
其中,所述治疗治疗耐药葡萄球菌感染性疾病的药物包括β-内酰胺类抗生素。
其中,所述的β-内酰胺类抗生素包括阿莫西林、头孢氨苄、头孢吡肟。
其中,它是由下述重量配比的原料药制备而成的制剂:
柠檬醛0.01-0.99份,β-内酰胺类抗生素0.01-0.99份。
本发明还提供了前述的药物在制备治疗耐药葡萄球菌感染性疾病的药物中的用途;优选地,所述耐药葡萄球菌感染性疾病为耐甲氧西林金黄色葡萄球菌感染性疾病;更优选地,所述的耐甲氧西林金黄色葡萄球菌感染性疾病为耐甲氧西林金黄色葡萄球菌ATCC43300或耐甲氧西林金黄色葡萄球菌ATCC33591感染性疾病。
本发明最后提供了前述的联合用药物在制备治疗耐药葡萄球菌感染性疾病的药物中的用途;优选地,所述耐药葡萄球菌感染性疾病为耐甲氧西林金黄色葡萄球菌感染性疾病;更优选地,所述的耐甲氧西林金黄色葡萄球菌感染性疾病为耐甲氧西林金黄色葡萄球菌ATCC43300或耐甲氧西林金黄色葡萄球菌ATCC33591感染性疾病。
本发明柠檬醛或其衍生物为天然药物,单用能有效抑制MRSA的生长或杀灭MRSA,能缓解MRSA感染引起的氧化应激及炎症反应,可用于治疗葡萄球菌感染性疾病;同时联合β-内酰胺类抗生素能显著增强抗MRSA的活性,具有良好的协同增效作用;将本发明药物制备成治疗MRSA感染药物,临床应用前景良好。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
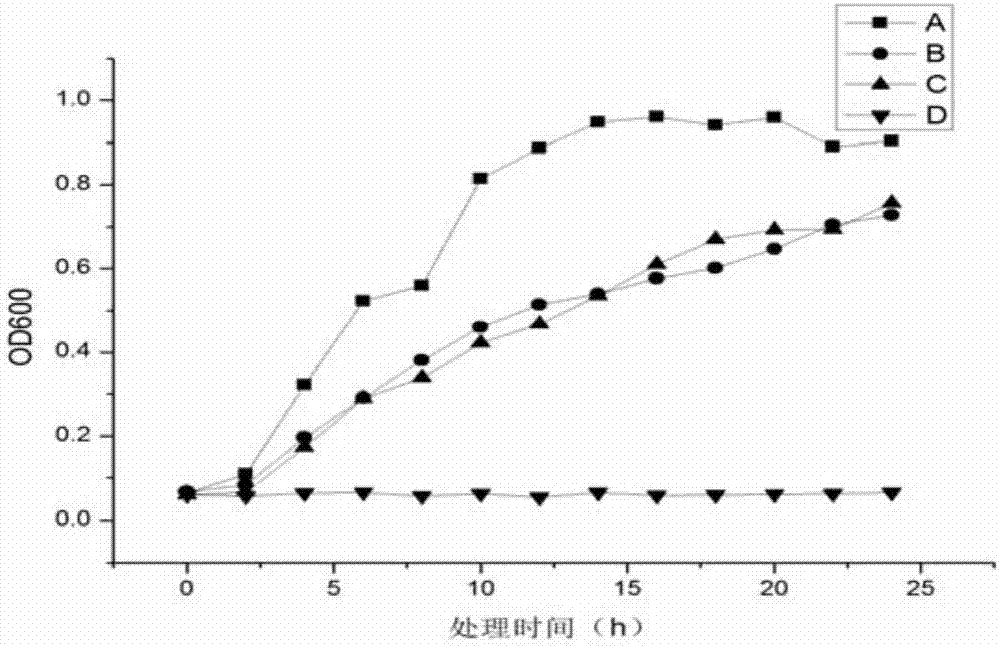
图1发明药对MRSA生长曲线的影响;注:A.空白对照,B.发明药1组(1/8MIC),C.发明药2组(1/4MIC),D.发明药3组(1/2MIC),E.发明药4组(3/4MIC);
图2发明药与阿莫西林联合使用对MRSA生长曲线的影响;注:A.空白组,B.发明药组(1/4MIC),C.阿莫西林组(1/4MIC),D.联合用药组(1/4MIC发明药+1/4MIC阿莫西林);
图3发明药对MRSA细胞超微结构的影响;注:A.空白对照组,B.溶剂对照组(Tween-80),C.发明药低剂量组(2MIC处理2h),D.发明药高剂量组(4MIC处理2h);
图4发明药对MRSA感染模型小鼠炎性细胞因子的影响;注:B.空白组,N.模型组,P.阳性药物组,H.发明药高剂量组,M.发明药中剂量组,L.发明药低剂量组;与空白组比较,*P<0.05,**P<0.01;与模型组比较,ΔP<0.05,ΔΔP<0.01;与阳性组比较,#P<0.05,##P<0.01
图5发明药对MRSA感染模型小鼠氧化因子的影响;注:B.空白组,N.模型组,P.阳性药物组,H.发明药高剂量组,M.发明药中剂量组,L.发明药低剂量组;与空白组比较,*P<0.05,**P<0.01;与模型组比较,ΔP<0.05,ΔΔP<0.01;与阳性组比较,#P<0.05,##P<0.01
图6 MRSA感染小鼠肺组织的病理变化;注:a.空白组,b.模型组;
图7 MRSA感染小鼠肝组织的病理变化;注:a.空白组,b.模型组;
图8 MRSA感染小鼠肾组织的病理变化;注:a.空白组,b.模型组;
图9发明药对MRSA感染小鼠肺组织的病理影响;注:a.空白组,b.模型组,c.阳性药物组,d.发明药组。
具体实施方式
实施例1柠檬醛及与β-内酰胺类抗生素联用体外抗MRSA活性研究
1实验材料
1.1实验菌株
标准株:耐甲氧西林金黄色葡萄球菌(methicillin resistant staphylococci aureus,MRSA)标准株ATCC43300和ATCC33591,购自美国典型菌种保藏中心;
临床分离株:22株,来源于四川省妇幼保健院,经VITEK32或16SrRNA鉴定为金黄色葡萄球菌;经头孢西丁抗菌药物敏感试验和mecA基因鉴定为MRSA,菌株来源和编号见表1。
表1:受试菌株的编号和来源
1.2药物
实验药物:发明药柠檬醛(Citral),Sigma-aldrich,批号MKBJ9477V。
抗生素:阿莫西林(Amoxicillin hydrochloride trihydrate),批号B326BA3634,生工生物工程(上海)股份有限公司;头孢氨苄(Cephalexin monohydrate),批号BA14BA0016,生工生物工程(上海)股份有限公司;头孢吡肟(cefepime),批号RK9Y-DN25,中国食品药品检定研究院。
1.3培养基
MUELLER-HINTON BROTH(MHB),批号583507,英国OXOID公司;MUELLER-HINTON AGAR(MHA),批号1376993,英国OXOID公司;营养琼脂,批号20150810,北京奥博星生物技术有限责任公司。
1.4主要试剂
Tween-80,批号20150429,国药集团化学试剂有限公司;麦氏比浊管,bioMerieux SA公司。
1.5主要仪器
生物安全柜(BIOsafe12),上海力申科学仪器有限公司;全自动高压灭菌锅(HICLAVE HVE-50),HIRAYAMA公司;隔水式恒温培养箱,上海一恒科学仪器有限公司;电热鼓风干燥箱(GZX-9240MBE),上海博迅实业有限公司医疗设备厂;优普UPH-II-10T纯水系统;分析天平(ME104),METTLER TOLEDO公司;Varioskan Flash光谱扫描多模板读数器,Thermo Fisher Scientific公司;BA200Digital数码三目摄像显微摄像系统,麦克奥迪实业集团有限公司;Tecnai G2F20,FEI公司。
2实验方法
2.1发明药体外抗MRSA活性
2.1.1菌液制备
活化各受试菌,挑单克隆菌落于0.9%生理盐水中,将菌液配置成0.5麦氏浓度(1.5×108CFU/ml)备用。
2.1.2药液配制
以Tween-80为乳化剂,制备发明药;以无菌水为溶剂,将抗生素配制成浓度为4096μg/ml的母液,4℃冰箱保存备用。采用二倍稀释法分别对发明药和抗生素母液进行系列稀释,共稀释成12个不同浓度梯度的稀释液,用于测定MIC、MBC和FIC。
2.1.3最低抑菌浓度(minimal inhibitory concentration,MIC)测定
微量稀释法测定发明药和β-内酰胺类抗生素(阿莫西林、头孢氨苄和头孢吡肟)的MIC。具体方法如下:在96-孔板每孔中依次加入MHB培养液、稀释药液和受试菌液,使发明药在每孔中的终浓度为43.9mg/ml~0.021mg/ml,抗生素的终浓度为2048μg/ml~1μg/ml,菌液的终浓度为1.5×106CFU/ml,37℃恒温培养18h,观察受试菌的生长情况,以抑制受试菌生长的最低药物浓度为该菌的MIC值。以不加药物为受试菌阳性对照,以不加菌液为药物阴性对照,以仅含培养液不含药液和菌液的为空白对照。每株受试菌进行三个平行实验,且实验重复三次。
2.1.4最低杀菌浓度(minimum bactericidal concentrations,MBC)测定
药液稀释和菌液配制同MIC测定。
MBC测定:按照MIC值测定方法,将培养基、受试菌株及受试药物加入96-孔板中,37℃培养18h,将未见细菌生长培养孔中的肉汤接种于MHA琼脂平板上,37℃培养18h,观察生长情况,以未见菌落生长的最低药物浓度为该药物的MBC。以不加药物为受试菌阳性对照,以不加菌液为药物阴性对照,以仅含培养液不含空白药液和菌液的为空白对照。每株受试菌进行三个平行实验,且实验重复三次。
2.2发明药与β-内酰胺类抗生素联合使用抗MRSA活性
药液稀释和菌液制备同MIC测定。
部分抑菌浓度(fractional inhibitory concentration,FIC)指数测定:根据发明药(甲药)和β-内酰胺类抗生素(以阿莫西林、头孢氨苄和头孢吡肟为代表药)(乙药)的MIC,将甲药和乙药分别进行倍比稀释,使其终浓度为2MIC~1/16MIC,棋盘法测定甲、乙两药联合用药的MIC。即在96孔板中分别加入甲药、乙药和受试菌液,37℃培养18h,观察结果,判定甲、乙两药联合用药的MIC。以不加药物为受试菌阳性对照,以不加菌液为药物阴性对照,以仅含培养液不含药物和菌液的为空白对照。每株受试菌进行三个平行实验,实验重复三次。
数据统计与分析:FIC=甲药联用的MIC/甲药单用的MIC+乙药联用的MIC/乙药单用的MIC)。其中FIC≤0.5为协同作用,0.5<FIC≤1为相加作用,1<FIC≤2为无关作用,FIC>2为拮抗作用。
3实验结果
3.1发明药体外抗MRSA活性
微量稀释法分别测定了发明药和β-内酰胺类抗生素(阿莫西林、头孢氨苄和头孢吡肟)对受试菌的MIC和MBC,结果见表2。
表2:发明药体外抗MRSA活性
Unit:μg/ml
由上表可知,临床MRSA分离株对常用3种β-内酰胺类抗生素(阿莫西林、头孢氨苄、头孢吡肟)均耐药,MIC值分别为4μg/ml~256μg/ml、16μg/ml~1024μg/ml、4μg/ml~1024μg/ml,MBC分别为128μg/ml~1024μg/ml、128μg/ml~1024μg/ml、64μg/ml~1024μg/ml。发明药体外具有较强抗MRSA活性,MIC为695μg/ml~2780μg/ml,MBC为1390μg/ml~2780μg/ml。MIC集中分布在695μg/ml~1390μg/ml,其MIC50和MIC90分别为758μg/ml和1122μg/ml。
3.2发明药增强β-内酰胺类抗生素抗MRSA体外活性
棋盘法测定了发明药与β-内酰胺类抗生素联合使用体外抗MRSA活性,统计分析FIC指数及其相互作用,结果见表3;统计分析β-内酰胺类抗生素单用和联合发明药使用MIC50和MIC90的变化,结果见表4。
表3:发明药与β-内酰胺类抗生素联合使用的相互作用
由表3可知,发明药与3种β-内酰胺类抗生素联合使用时,具有协同增效的效果,例如:对耐甲氧西林金黄色葡萄球菌ATCC43300和ATCC33591有协同作用。
表4:发明药增强β-内酰胺类抗生素活性
Unit:μg/ml
由表4可知,发明药与β-内酰胺类抗生素(阿莫西林、头孢氨苄、头孢吡肟)联合使用可明显降低抗生素对MRSA的MIC50和MIC90。如阿莫西林的MIC50由单用的24.05μg/ml降低为1.25μg/ml,降低了19.27倍,头孢氨苄和头孢吡肟的MIC50分别降低了11.9倍和16.22倍;阿莫西林、头孢氨苄和头孢吡肟的MIC90则分别降低了13.98倍、12.38倍和14.94倍。说明发明药可有效降低β-内酰胺类抗生素在防治MRSA感染性疾病中的用量,具有增强β-内酰胺类抗生素抗MRSA感染活性。
由上述实验结果可知:发明药柠檬醛既有抗MRSA体外活性,还有增强β-内酰胺类抗生素抗MRSA活性。低浓度发明药可延长MRSA生长迟缓期,降低细菌生长总量,呈抑制作用;高浓度发明药可破坏细胞超微结构(细胞膜、细胞壁和胞质),出现胞质内容物渗漏,呈杀灭作用;其抑制和杀灭作用均呈明显量效关系。
实施例2柠檬醛体内抗MRSA感染疗效研究
1实验材料
1.1实验菌株
标准株:耐甲氧西林金黄色葡萄球菌(methicillin resistant staphylococci aureus,MRSA)标准株ATCC43300,购自美国典型菌种保藏中心;
1.2药物
实验药物:发明药柠檬醛(Citral),Sigma-aldrich,批号MKBJ9477V。
阳性药物:Vancomycin,Sigma公司。
1.3实验动物
KM小鼠,SPF级,雌雄各半,体重(20±2)g,四川省成都生物制品研究所有限责任公司提供,动物生产许可证号SCXK(川)2016-08。
1.4培养基
MUELLER-HINTON BROTH(MHB),批号583507,英国OXOID公司;MUELLER-HINTON AGAR(MHA),批号1376993,英国OXOID公司;营养琼脂,批号20150810,北京奥博星生物技术有限责任公司。
2实验方法
2.1MRSA感染小鼠模型制备
MRSA毒力测定:KM小鼠(SPF级)适应性饲喂后,按体重随机分为空白组和5个实验组,每组10只。MRSA标准株ATCC43300 37℃恒温培养至对数期,用生理盐水将菌液稀释成五个不同浓度梯度,腹腔注射小鼠。在空白组小鼠不死亡的前提下,记录各实验组小鼠72h内的死亡率,测定SPF小鼠全部死亡的最低细菌量,即为ATCC43300的最小全死量(Minimum lethal dose,MLD)。
MRSA全身感染模型小鼠制备:以MRSA的MLD菌液腹腔注射KM小鼠(SPF级),制备MRSA感染小鼠模型。
2.2实验分组及给药
肌内注射:KM小鼠(SPF级)适应性饲喂后,按体重随机分为11组,即空白组,模型组,阳性组,实验1、2、3、4、5、6、7、8共8个剂量组,每组10只。其中空白组和模型组肌内注射生理盐水,阳性组肌内注射万古霉素,实验组分别按不同剂量注射发明药(给药剂量详见表5),1次/d,连续给药3d。除空白组不感染MRSA外,其余各组小鼠均在给药第3d后,用MRSA的MLD菌液量腹腔注射小鼠,观察小鼠攻毒感染后7d的存活情况。
灌胃:KM小鼠(SPF级)适应性饲喂后,按体重随机分为11组,即空白组,模型组,阳性组,实验1、2、3、4、5、6、7、8共8个剂量组,每组小鼠10只。其中空白组和模型组灌胃生理盐水,阳性组灌胃万古霉素,实验组分别按不同剂量灌胃发明药(给药剂量详见表6),1次/d,连续给药3d。除空白组不感染MRSA外,其余各组小鼠均在第3d给药后,用MRSA的MLD菌液量腹腔注射小鼠,观察小鼠攻毒感染后7d的存活情况。
数据统计与分析:统计小鼠攻毒感染后7d内的存活情况,分析发明药对MRSA感染模型小鼠的体内保护率;寇氏法统计分析发明药肌内注射和灌胃给药的半数有效量(50%effective dose,ED50)。
3实验结果
3.1肌内注射抗MRSA感染疗效
采用预防给药方式,用发明药肌内注射小鼠,1次/d,连续给药3d,最后一次给药后用MRSA腹腔注射小鼠,观察小鼠感染后7d的存活情况,结果见表5。
表5:发明药肌内注射抗MRSA感染疗效
由表5可知,发明药肌内注射预防给药,对由MRSA诱导感染模型小鼠有很好的体内抗感染疗效,并呈明显量效关系,具有与阳性药物万古霉素相当的作用。其中高剂量(0.36g/kg)的体内保护率达100%;寇氏法统计分析,发明药肌内注射抗MRSA感染的ED50为0.08g/kg。
3.2发明药灌胃抗MRSA感染疗效
采用预防给药方式,用发明药灌胃小鼠,1次/d,连续给药3d,最后一次给药后用MRSA腹腔注射小鼠,观察小鼠感染后7d的存活情况,结果见表6。
表6:发明药灌胃抗MRSA感染疗效
由上表可知,发明药灌胃,对MRSA感染模型小鼠有明显的抗感染疗效,呈明显量效关系,具有与阳性药物万古霉素相似的作用。其中1.26g/kg发明药灌胃的体内保护率达100%;寇氏法统计分析,发明药灌胃对MRSA感染模型小鼠的ED50为0.25g/kg。
由上述实验结果可知:柠檬醛采用肌内注射和灌胃2种预防给药方式,均有体内抗MRSA感染疗效,呈明显量效关系,ED50分别为0.08g/kg和0.25g/kg。
实施例3柠檬醛及与β-内酰胺类抗生素联用抗MRSA的作用机制
1实验材料
1.1实验菌株
标准株:耐甲氧西林金黄色葡萄球菌(methicillin resistant staphylococci aureus,MRSA)标准株ATCC43300,购自美国典型菌种保藏中心;
1.2药物
实验药物:发明药柠檬醛(Citral),Sigma-aldrich,批号MKBJ9477V。
抗生素:阿莫西林(Amoxicillin hydrochloride trihydrate),批号B326BA3634,生工生物工程(上海)股份有限公司;Vancomycin,Sigma公司。。
1.3实验动物
KM小鼠,SPF级,雌雄各半,体重(20±2)g,四川省成都生物制品研究所有限责任公司提供,动物生产许可证号SCXK(川)2016-08。
1.4培养基
MUELLER-HINTON BROTH(MHB),批号583507,英国OXOID公司;MUELLER-HINTON AGAR(MHA),批号1376993,英国OXOID公司;营养琼脂,批号20150810,北京奥博星生物技术有限责任公司。
1.5主要试剂
Mouse TNF-α(Tumor Necrosis Factor Alpha)ELISA kit.96T,批号AK0017MAY19010,Elabscience公司;Mouse IL-1β(Inter leukin 1Beta)ELISA kit.96T,批号AK0017MAY19012,Elabscience公司;Mouse IL-6(Inter leukin6)ELISA kit.96T,批号AK0017MAY19011,Elabscience公司。SOD试剂盒(WST-1法),批号20170518,南京建成生物工程研究所;谷胱甘肽过氧化物酶(GSH-Px)试剂盒,批号20170515,南京建成生物工程研究所;丙二醛(MDA)试剂盒,批号20170517,南京建成生物工程研究所;羟基自由基(·OH)试剂盒,批号20170516,南京建成生物工程研究所。0.9%氯化钠注射液,批号B16051903,四川科伦药业股份有限公司。
2实验方法
2.1体外作用机制
2.1.1发明药对MRSA生长曲线的影响
取ATCC43300对数期菌液,将菌液浓度调至1.5×108CFU/ml备用。在MHB培养基中分别加入MRSA菌液和发明药(1/8MIC、1/4MIC、1/2MIC、3/4MIC),每隔2h取样,检测OD600;以不加药物为空白对照。以OD600为纵坐标,时间为横坐标,EXCEL软件绘制MRSA生长曲线。
2.1.2发明药与阿莫西林联合使用对MRSA生长曲线的影响
β-内酰胺类抗生素以阿莫西林为代表,分析发明药与抗生素联用对MRSA生长曲线的影响。取ATCC43300对数期菌液,将菌液浓度调至1.5×108CFU/ml备用。在MHB培养基中分别加入MRSA菌液和药物(1/4MIC发明药,或1/4MIC阿莫西林,或1/4MIC发明药+1/4MIC阿莫西林),每隔2h无菌取样,检测OD600;以不加药物为空白对照。以OD600为纵坐标,时间为横坐标,EXCEL软件绘制MRSA的生长曲线。
2.1.3发明药对MRSA超微结构的影响
药物处理:取MRSA对数期菌液,分别用2MIC发明药处理2h,4MIC发明药处理2h,分析发明药浓度和作用时间对MRSA超微结构的影响;以不加药物为空白对照,Tween-80为溶剂对照。
菌液固定:取处理后的菌液10000r/min离心10min,弃上清,PBS洗涤3次;加入0.5%戊二醛固定液,4℃静置10min进行预固定;10000r/min离心15min,弃上清,加入3%戊二醛固定液进行固定。
样品制备及超微结构分析:采用锇酸后固定,醋酸铀块染,丙酮包埋后超薄切片,用Tecnai G2F20电子显微镜观察MRSA超微结构的,由四川大学分析测试中心完成。
2.2体内作用机制
2.2.1发明药对MRSA感染模型小鼠细胞因子和氧化因子的影响
用发明药肌内注射,以0.36g/kg、0.23g/kg、0.15g/kg为高剂量(H)、中剂量(M)和低剂量(L)组,比较分析发明药对MRSA感染模型小鼠治疗前后炎性细胞因子和氧化因子的变化,并分析量效关系。
血清制备:发明药预防给药3d,用MRSA攻毒感染小鼠,感染7d后,对各组存活小鼠进行眼眶取血,制备血清。
炎性细胞因子测定:ELISA法测定小鼠血清中主要炎性细胞因子TNF-α、IL-6和IL-1β的含量。
氧化因子测定:ELISA法测定小鼠血清中谷胱甘肽过氧化物酶(GSH-Px)、超氧化物歧化酶(SOD)、丙二醛(MDA)和羟基自由基(·OH)含量。
数据统计与分析:用SPSS 19.0软件单因素方差分析法,比较分析空白组、模型组、阳性组和发明药组小鼠体内炎性细胞因子和氧化因子的差异,其中P<0.05表示具有统计学差异,P<0.01表示具有显著统计学差异。
2.2.2发明药对MRSA感染模型小鼠病理变化的影响
用发明药肌内注射预防给药,选择高剂量组(0.36g/kg)为实验组,分析发明药高剂量作用下对MRSA感染模型小鼠组织病理变化的影响。
样品采集和固定:发明药肌内注射预防给药3d,用MRSA感染,取感染7d后小鼠的肺、肝和肾,生理盐水洗涤后放入4%甲醛固定液进行固定,24h后更换固定液,保存于4℃冰箱,用于病理切片制作。以不给药物不感染的为空白组,以不给药只感染的为模型组,以给药万古霉素为阳性组。
制备病理切片和病理分析:对固定好的肺、肝和肾进行脱水处理,后包埋、切片和染色,进行显微图像采集和病理切片分析,由成都里来生物科技有限公司完成。
3实验结果
3.1体外作用机制
3.1.1发明药对MRSA生长的影响
生长曲线法分析发明药体外对MRSA标准株生长的影响,结果见图1。
由图1可知,发明药能明显延长MRSA的迟缓期,抑制MRSA生长,细菌生长量明显降低,当发明药浓度增加到3/4MIC时,MRSA生长完全处于被抑制状态。发明药抑制MRSA生长呈明显量效关系,与“体内抗感染疗效结果”一致。
3.1.2发明药与阿莫西林联合使用对MRSA生长的影响
以阿莫西林为β-内酰胺类抗生素代表,分析发明药、阿莫西林、发明药和阿莫西林联合使用对MRSA生长曲线的影响,结果见图2。
由上图可知,与空白组比较,发明药(1/4MIC)和阿莫西林单独使用,一定程度上能抑制MRSA生长及其生长总量;而发明药(1/4MIC)与阿莫西林(1/4MIC)联合使用时,抑制作用显著高于两者相同浓度的单独作用,生长完全被抑制,呈现明显协同增效作用,与“发明药具有自身抗MRSA体外活性和协同增效β-内酰胺类抗生素抗MRSA体外活性”结果一致。
3.1.3发明药对MRSA细胞超微结构的影响
将ATCC43300培养至对数期,分别用2MIC发明药处理2h和4MIC发明药处理2h,以不加药物的为空白对照,Tween-80为溶剂对照。透射电镜观察其超微结构变化,结果见图3。
由图3可知,空白组和溶剂组的MRSA细胞结构完整,细胞分裂正常。2MIC发明药处理2h时,MRSA细胞结构相对较完整,部分细胞膜受到损坏,细胞壁和细胞膜界限模糊;4MIC发明药处理2h时,MRSA细胞结构完全被破坏,细胞壁和细胞膜脱落,胞质内含物被分解,胞内容渗漏,细胞碎片增多。说明破坏MRSA超微结构主要与发明药的浓度有关,即发明药在低浓度作用下对MRSA呈抑制作用,在高浓度作用下呈杀伤作用。
3.2体内作用机制
3.2.1发明药对MRSA感染小鼠炎性细胞因子和氧化因子的影响
比较分析发明药对MRSA感染小鼠炎性细胞因子(图3)和氧化因子(图4)的影响。
由图4可知,MRSA感染后,模型组小鼠3种炎性细胞因子(IL-1β、IL-6和TNF-α)的含量均显著高于空白组(P<0.01);经发明药治疗后,3种炎性细胞因子含量均明显下降,并呈量效关系。与空白组比较,阳性药物组的IL-6和TNF-α含量无统计学差异(P>0.05),恢复至正常水平;发明药高剂量组小鼠的IL-1β和TNF-α含量无统计学差异(P>0.05),恢复至正常水平。与模型组比较,阳性药物组的IL-1β和IL-6均显著降低(P<0.01)、TNF-α明显降低(P<0.05);发明药除低剂量组TNF-α含量降低不具有统计学差异(P>0.05),中剂量组TNF-α明显降低(P<0.05)外,其余各组小鼠的IL-1β、IL-6和TNF-α含量均显著降低(P<0.01)。与阳性药物组比较,发明药高剂量组TNF-α、中剂量组IL-1β无明显统计学差异(P>0.05)外,其余各组IL-1β、IL-6和TNF-α有明显统计学差异(P<0.05,P<0.01)。说明发明药能通过降低MRSA感染小鼠的IL-1β、IL-6和TNF-α含量以控制MRSA感染,具有与阳性药物万古霉素相似的作用。
由图5可知,MRSA感染后,模型组小鼠的氧化因子GSH-Px和SOD含量显著降低(P<0.01),MDA和·OH含量显著升高(P<0.01);经发明药治疗后,GSH-Px和SOD含量显著增加、MDA和·OH含量显著降低,呈明显量效关系。与空白组比较,阳性药物组GSH-Px和SOD含量无统计学差异(P>0.05),恢复至正常水平,MDA和·OH含量虽叫模型组明显降低,但仍显著高于空白组(P<0.01);发明药除中、低剂量组的GSH-Px含量无明显统计学差异(P>0.05),恢复至正常水平外,高剂量组的GSH-Px含量和高中低剂量组的SOD、MDA和·OH含量均有显著统计学差异(P<0.01,P<0.05)。与模型组比较,阳性药物组的GSH-Px、SOD、MDA和·OH含量有显著统计学差异(P<0.01);除发明药低剂量组的GSH-Px和·OH含量无明显统计学(P>0.05)、中剂量组的GSH-Px有明显统计学差异(P<0.05)外,发明药其余各组的GSH-Px、SOD、MDA和·OH含量均有显著统计学差异(P<0.01)。与阳性组比较,发明药高、中剂量组的GSH-Px,高剂量组的MDA和·OH含量无明显统计学差异(P>0.05)。说明发明药可通过调节MRSA感染模型小鼠氧化因子GSH-Px、SOD、MDA和·OH含量,以控制MRSA感染,与阳性药万古霉素具有相似作用。
3.2.2发明药对MRSA感染小鼠的病理影响
MRSA感染对小鼠的病理影响:用MLD的MRSA菌液量腹腔注射小鼠,以肺、肝和肾为代表,分析小鼠感染MRSA后内脏器官的组织病理变化。其中肺组织的病理变化见图6,肝组织的病理变化见图7,肾组织的病理变化见图8。
由图6、图7和图8可知,MRSA腹腔注射小鼠,主要引起小鼠肺组织的炎性病理变化,肺出现明显炎性变化,肺间质细胞内中性粒细胞大量浸润,少量肺泡腔塌陷;肝组织和肾组织均无明显病理变化。
发明药对MRSA感染小鼠肺组织的病理影响:用发明药高剂量(0.36g/kg)注射给药,分析发明药对MRSA感染模型小鼠肺组织病理变化的影响,结果见图9。
由图9可知,发明药治疗后,MRSA感染模型小鼠肺部炎性病理变化得到明显改善,肺间质细胞内无明显中性粒细胞浸润,肺泡上皮细胞无病变,肺泡结构清晰,支气管结构完整。说明发明药能抑制并改善肺部炎性病变,控制小鼠肺部炎症,以消除MRSA感染。
由上述结果可知:腹腔注射MRSA感染小鼠,主要引起小鼠肺组织病理变化,肝组织和肾组织无明显病理变化。发明药体内抗MRSA感染机制主要与其调节MRSA感染小鼠炎性细胞因子(IL-1β、IL-6和TNF-α)和氧化因子(GSH-Px、SOD、MDA和·OH)含量,改善肺组织炎性细胞浸润,修复炎性病变,调节氧化应激能力等因素有关。
综上,本发明柠檬醛或其衍生物为天然药物,单用能有效抑制MRSA的生长或杀灭MRSA,能缓解MRSA感染引起的氧化应激及炎症反应,可用于治疗葡萄球菌感染性疾病;同时联合β-内酰胺类抗生素能显著增强抗MRSA的活性,具有良好的协同增效作用;将本发明药物制备成治疗MRSA感染药物,临床应用前景良好。
- 还没有人留言评论。精彩留言会获得点赞!